



Volume 01 ,No.1 Pages 4 - 9
1. 最近の研究から/FROM LATEST RESEARCH
電解水素化反応の反応機構解明に向けた in situ XAFS 測定の活用
Application of In Situ XAFS Measurements for Mechanistic Elucidation of Electrocatalytic Hydrogenation Reactions
[1]横浜国立大学 大学院工学研究院 Department of Chemistry and Life Science, Yokohama National University、[2]京都大学 大学院工学研究科 Graduate School of Engineering, Kyoto University
- Abstract
- 本稿では、ピリジン類を代表とする窒素含有芳香族化合物の電解水素化反応に関する最新の研究成果を紹介し、その反応機構解明においてビームラインBL14B2で実施したin situ X線吸収微細構造(XAFS)測定が果たした役割を詳述する。アニオン交換膜型電解セルとRh触媒を用いることで、常温・常圧かつ酸添加剤を用いずに、ピペリジンを高効率かつ定量的に合成することに成功した。XAFS測定では、反応中におけるRh種の還元過程を可視化し、電解運転条件において活性種としてRh(0) が発生していることを示した。本成果は、触媒設計・反応場制御・解析技術が融合することで、電解合成の高度化が可能であることを示しており、SPring-8 の放射光測定がその基盤として大きく貢献している。
1. はじめに
21 世紀に入り、世界の産業構造はかつてない規模での転換を迫られている。その中心にあるのが脱炭素社会の実現という目標である。気候変動に対する国際的な合意、すなわちパリ協定に端を発した温室効果ガス排出削減の枠組みは、エネルギー・輸送・製造といった従来の重厚長大な産業分野だけでなく、化学産業にも大きな影響を与えている。
特に、化学産業はエネルギー集約型であることに加え、製品ライフサイクルの中でもっとも上流に位置する基礎化学品の製造過程において、大量の化石資源を原料および熱源として用いている。そのため、CO2排出量の全体に占める割合も無視できず、持続可能な未来に向けたプロセス革新が不可欠とされている。
このような背景から、近年注目されているのが、再生可能エネルギー由来の電力を直接駆動力とする電解合成プロセスである。電解合成は、反応を電位で制御し、電子そのものが試薬となるため、温度や圧力に依存しない温和な条件での選択的な分子変換を可能とする。とくに、熱的条件では制御が難しい高選択的還元・酸化反応や、フロー系を通じたスケーラブルな合成手法との親和性が高く、学術界・産業界を問わず関心が高まりつつある。
その中でも、芳香族化合物の電解水素化反応は、電気化学の新たな応用先として特筆すべき領域である。芳香環を部分的または完全に飽和させることで、分子構造を2次元から3次元へと変換することが可能であり、医薬品・農薬・香料・高機能材料などの合成において多面的な有用性を発揮する。
例えば、ピリジン類の水素化によって合成されるピペリジン類は、医薬品の極めて重要な構造単位であり、その選択的な水素化手法の開発は、学術的にも産業的にも大きな意義を有する。従来の方法では、高圧水素ガスや高温、酸添加剤を要し、環境負荷や装置の安全性、操作性の面で多くの制約があった。
我々の研究グループは、こうした制約を打破すべく、電気を用いた温和かつ選択的な芳香族水素化反応の開発に取り組んできた[1][1] N. Shida, Y. Shimizu, A. Yonezawa, J. Harada, Y.Furutani, Y. Muto, R. Kurihara, J. N. Kondo, E.Sato, K. Mitsudo, S. Suga, S. Iguchi, K. Kamiya, M. Atobe: J. Am. Chem. Soc. 146 (2024) 30212-30221.
。本稿では、近年報告したピリジン類の電解水素化に関する研究成果を起点として、SPring-8 におけるX線吸収微細構造(XAFS)測定が果たした役割と、今後の電解合成研究における展望について述べる。
2. ピリジン類の選択的水素化とその課題
ピリジンは、窒素原子を1つ含む6員環状の含窒素芳香族化合物である。その還元体であるピペリジンは、米国食品医薬品局(FDA)により承認されている低分子医薬品の約9%(2021年時点)に含まれている重要な化学構造である(図1a)[2, 3][2] E. Vitaku, D. T. Smith, J. T. Njardarson: J. Med. Chem. 57 (2014) 10257-10274.
[3] P. Bhutani, G. Joshi, N. Raja, N. Bachhav, P. K. Rajanna, H. Bhutani, A. T. Paul, R. Kumar: J. Med. Chem. 64 (2021) 2339-2381.。ピリジン環の核水素化、すなわち6e–/6H+ の導入による芳香環の還元は、ピペリジン骨格を構築する最も直線的な合成法であり、化学的な変換法が様々開発されてきた。特に工業的に有用性の高い不均一触媒と水素ガスを用いる核水素化反応は、古くから研究されてきた(図1b)。ピリジンをルイス酸やプロトン酸によって活性化することで比較的温和な条件で水素化を行うことができるが、これは資材コストの増加や後工程の煩雑化につながるため、添加剤無しの水素化反応が望ましい。しかし、添加剤を用いない水素化の多くが高圧(20–60 atm)および高温(30–160 ℃)条件によって反応を駆動している[4-6][4] F. Chen, W. Li, B. Sahoo, C. Kreyenschulte, G. Agostini, H. Lund, K. Junge, M. Beller: Angew. Chem. Weinheim Bergstr. Ger. 130 (2018) 14696-14700.
[5] W. Qian, L. Lin, Y. Qiao, X. Zhao, Z. Xu, H. Gong, D. Li, M. Chen, R. Huang, Z. Hou: Appl. Catal. A Gen. 585 (2019) 117183.
[6] F. Martinez-Espinar, P. Blondeau, P. Nolis, B. Chaudret, C. Claver, S. Castillón, C. Godard: J. Catal. 354 (2017) 113-127.。
このような文脈において、電解水素化は極めて有望な選択肢と考えられる。電気エネルギーを直接反応駆動力とするため、熱源としての化石燃料への依存を回避できるほか、反応場を細やかに制御できるという利点がある。特に、再生可能電力と連動させることで、CO2排出ゼロの分子変換を実現するポテンシャルを持つ。
実際、電気化学的なピリジン類の水素化反応は100年以上前にも研究が行われているが[7][7] J. G. Keay: Comprehensive Organic Synthesis, Elsevier. (1991) 579-602.
、意外なことに現在に至るまで効率的かつ選択的に、ピリジン類をピペリジン類へと変換する電気化学な手法は存在しなかった。この原因として、一般的な電解反応では電極からピリジン環に電子が注入されるステップから反応が開始するが、ここで形成されるラジカルアニオン種が極めて不安定であるため、プロトン化を受ける前に2量化などの副反応が起こってしまう(図1c)。このように電子注入とプロトン付加が段階的に進行する反応機構において、6e–/6H+ の受け渡しが必要となるピリジン→ピペリジンの変換は極めて難易度が高いと言える。
本研究では、アニオン交換膜(AEM)型リアクターを活用し、炭素担持Rh触媒(Rh/KB)を用いることで、ピリジンからピペリジンへの6電子還元反応を、常温・常圧・酸添加剤なしで実現した(図1d)。本研究で用いたAEM型リアクターにおいては、金属触媒表面に発生する水素化性種(Hads)がピリジンに付加することで、電子とプロトンが同時に受け渡しされ、不安定なラジカルアニオン中間体の形成を回避することができ、これによって効率的な反応が実現されている。加えて、本研究の大きな特長は、in situ XAFS測定によって、触媒表面におけるRh種の還元過程を追跡した点にある。このように、反応場の設計とその場構造解析を組み合わせた戦略こそが、持続可能な分子変換の実現に向けた鍵であると考えている。
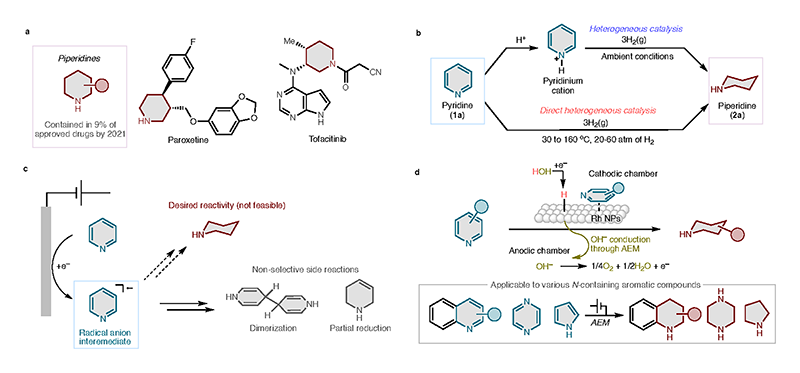
図1.本研究の背景。(a) ピペリジン骨格を有する医薬品の例、(b) 不均一触媒を用いるピリジン類の水素化、(c) 従来的な電解反応を利用するピリジン類の電解水素化とその課題、(d) 本研究の概要。文献1より転載。
3. 本研究の概要と主な成果
本研究では、窒素含有芳香族化合物の代表であるピリジン誘導体に対して、常温・常圧かつ酸添加剤を一切用いない条件下で、定量的かつ高選択的に水素化反応を進行させることを目的とした。そのための中核的な技術基盤として、AEM型リアクターを設計・構築した(図2a)。
本電解セルの特長は、従来のバッチセルとは異なり、陽極と陰極が固体高分子膜(ここではトクヤマ製A201膜)によって密着している点にある。この構造により、膜内部でのイオン輸送により回路が成立するため、電解質溶液中に支持電解質を添加する必要がない。さらに、反応液がポンプによって電解セル中に送液されるフロー型電解であるため、スケーラビリティに優れ、実用性が高い。
触媒には、ロジウム(Rh)をケッチェンブラック(KB)に担持したRh/KB触媒を最適とした。電極はカーボンペーパーを担体とし、触媒インクをスプレー塗布することで電極(Catalyst-Coatedsubstrate, CCS)を作製した。実験では、基質としてピリジン(1.0 mmol)を含む水溶液を陰極側電解液として用いた。陽極では、陰極で生成したOH–が酸化され酸素を発生する反応が進行するため、特に電解液を流通させる必要はなく、大気に解放している。電解は定電流条件下で行い、通電量(Fmol–1)と生成物収率の相関を評価し、電流効率として算出した。結果として、Rh/KB触媒を用いた系では、99%以上の収率でほぼ定量的にピペリジンが得られ、副生成物が一切検出されないことを確認した。一方、本反応の成否は電極触媒の金属種に強く依存することを見出しており、例えば同じ白金族であるプラチナ(Pt)を用いた場合にはほとんど目的の反応は進行しなかった。
特筆すべきは、反応後期に至るまで、電流効率がほぼ100%に近いという点である(図2b)。すなわち、投入した電気エネルギーのほとんどが目的生成物の形成に寄与しており、副反応としての水素発生反応(HER)は極めて抑制されている。この結果は、電極表面での選択的吸着と脱離のバランスが良好であることによって、選択性を発揮していることを示唆する。
さらに本反応系は、ピリジンのみならず、他の含窒素芳香族化合物にも適用可能であることを実証した。たとえば、キノリン、ピラジン、さらには電子豊富な含窒素芳香族化合物であるピロールにも拡張でき、それぞれにおいて高い変換率と収率、選択性が確認された(図1d)。
これらの成果は、従来の熱的還元反応に比べ、はるかに温和な条件下で、高付加価値の化合物群を効率よく合成可能であることを実証したものである。さらに、このような反応系を理解し制御するためには、触媒表面で進行する電子状態や中間体の挙動をその場観測により可視化する必要がある。次節では、その鍵となったin situ XAFS測定について詳述する。
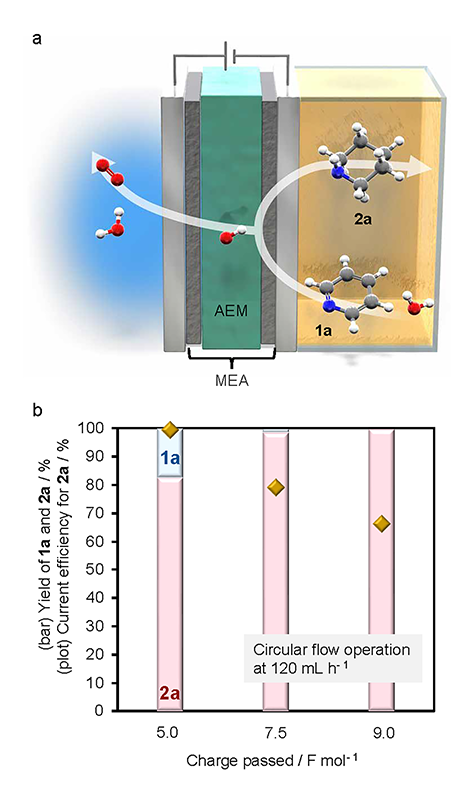
図2.ピリジンの電解水素化 (a) AEM型リアクターを用いた反応系の概略、(b) 定電流電解におけるピペリジン生成量と電流効率の通電量依存性。文献1より一部改変し掲載。
4. 機構解明における in situ XAFS測定の意義
電解反応の最適化において、反応中における触媒の電子状態や構造変化を把握することは極めて重要である。とりわけ、遷移金属を担体に分散させたナノ粒子触媒の場合、表面の酸化還元状態、配位環境、粒子サイズ、集合状態などが反応性に大きな影響を与えることが知られている。しかしながら、これらの情報は従来のex situ分析手法では把握が困難であり、反応中の真の活性状態を捉えるためには、in situ(またはoperando)測定によるリアルタイム観測が不可欠である。
本研究においては、SPring-8 のBL14B2ビームラインにおいて、in situ XAFS測定を実施した。特に、XANES領域のスペクトル変化から、反応条件におけるRh種の電子状態を観測することを目的とした。
測定セルの設計
in situ測定には、X線窓を有しながら電解反応の効率を損なわない特殊なセル設計が求められる。本研究では、我々が以前に開発したin situ赤外分光(IR)測定用レーザー透過型電解セルの設計思想を継承し、X線に対しても高い透過性を有する材料構成へと最適化を行った。IR測定用セルは、我々の研究グループにおいて、固体高分子電解質電解環境下におけるHadsの直接観測[8][8] J. N. Kondo, S. Ge, T. Suzuki, R. Osuga, T. Matsumoto, T. Yokoi, Y. Shimizu, A. Fukazawa, N. Shida, M. Atobe: J. Phys. Chem. C Nanomater. Interfaces. 126 (2022) 19376-19385.
、ならびに中間体の吸着様式を可視化する目的で開発されたものである[9][9] Y. Shimizu, J. Harada, A. Fukazawa, T. Suzuki, J. N. Kondo, N. Shida, M. Atobe: ACS Energy Lett. (2023) 1010-1017.
。
その基本構造は、中央に高分子電解質膜を挟み、両側に触媒層を担持した電極を密着させ、レーザーやX線が通過する小型窓を配置したものである(図3a)。これにより、通電しながら任意の波長の放射光を電極界面に照射し、反応中の構造変化をリアルタイムで検出できる。IR測定においてはCaF2 窓を用いたが、XAFS測定にあたりポリイミドフィルムを窓材として使用し、電解とin situ測定の両立を実現した。また,測定対象であるカソードのRh K端の測定を妨げないよう、アノードとして酸素発生の触媒能を有するDSE© 電極を用いた。本セットアップにより、透過法によるin situ XAFS測定が可能となった(図3b)。
今回のXAFS測定では、このセルにRh/KB触媒を担持したカーボンペーパーを取り付け、反応条件下(定電流電解)におけるRh K端XANESスペクトルの変化を追跡した。
XAFSによる酸化状態変化の可視化と構造解析
XANESスペクトルの測定により、反応開始前のRh種にはRh(0) とRh(III) が混在していることが示された。これは、触媒調製および電極作製後の空気酸化によって特にナノ粒子表面に酸化物層が形成されていたためと考えられる。通電を開始すると、時間とともにXANESスペクトルに変化が現れ、Rh K端の吸収端位置が低エネルギー側へシフトするとともに、ホワイトライン強度が低下した。よって、電気化学的にRh種が逐次的にRh(0) へと還元されていくことが明らかとなった(図3c)。

図3.in situ XAFS測定。(a) in situ XAFS測定に用いた電解セルの概観、(b) BL14B2において構築した実験のセットアップの写真、 (c) 得られたRh K端XANESスペクトル。文献1より一部改変し掲載。
計算科学への展開と機構解析
Rh(0) が反応における触媒活性を有するというin situ XAFS測定の知見に基づき、密度汎関数(DFT)計算を実施した。吸着→電解水素化→脱着の過程をそれぞれシミュレーションした。Rh(0) 上での水素化反応のエネルギーダイアグラムを解析したところ、生成物であるピペリジンの脱離過程に最も多くのエネルギーが必要となっており、この段階が律速段階であることが示唆された。また、触媒活性をほとんど示さなかったPt においても脱着過程のエネルギー計算を行うと、Pt はピペリジンの脱着により大きなエネルギーを要することも示唆された。このように、in situ XAFS測定によって反応系中でRh(0) が生成することを実証したことで、DFT計算における正確なモデルを選択することが可能となり、触媒の金属種が反応のどのステップに影響を与え、反応速度や収率に違いを与えたのかを明らかとするに至った。
合金電極触媒の解析
さらに我々は、Ni・Co・Fe系の非貴金属触媒を用いたAEM型リアクターにおけるアルコール酸化反応の検討を行い、その過程でXAFS測定を活用した触媒の電子状態評価を行っている[10][10] Y. Furutani, Y. Shimizu, J. Harada, Y. Muto, A. Yonezawa, S. Iguchi, N. Shida, M. Atobe: ACS Catal. 14 (2024) 8922-8929.
。本研究ではin situ 測定ではないものの、NiFe やNiCo合金触媒のXANESおよびEXAFS解析を通じて、構成元素の混合状態や酸化還元挙動の違いを明らかにした。
このようなXAFSによる電子状態・局所構造の解析は、触媒活性点の設計指針となり、非貴金属を用いた高選択的反応系の構築に貢献している。
5. おわりに
本稿では、我々の研究グループが近年発表したピリジン類の電解水素化反応に関する研究成果を軸に、SPring-8でのin situ XAFS測定が果たした役割と、そこから得られた知見について紹介した。電解反応という、動的な反応場における触媒の構造や電子状態を、リアルタイムかつ高精度で捉える手段として、放射光XAFSは極めて有効であり、反応機構の定量的解明にとって不可欠な分析技術となっている。また、in situ測定のみならず、静的条件下でのXAFS解析も材料開発や構造–性能相関の確立に大きく貢献しうることを示した。
SPring-8という世界有数の放射光施設は、物質の深部構造と反応の本質を“見る”ための窓であり、その可能性は無限と言える。今後も、電気化学と放射光科学が緊密に連携し、エネルギー・物質変換・環境のあらゆる分野において新たな価値を創出していくことを、我々は確信している。
本稿に掲載したXAFS測定は、すべてSPring-8のBL14B2において実施したものです。測定にあたり、公益財団法人高輝度光科学研究センター 主幹研究員 渡辺剛 博士には、反応系の設計から組み立てまで、大変なご尽力を賜りました。この場をお借りして厚く御礼申し上げます。
本研究を実施するにあたり、AEMおよびアイオノマーは株式会社トクヤマのご厚意により提供されたものです。また、本稿に示した研究成果は、多くの共同研究者のご尽力により発表に至ったものです。関係各位に厚くお礼申し上げます。
参考文献
[1] N. Shida, Y. Shimizu, A. Yonezawa, J. Harada, Y.Furutani, Y. Muto, R. Kurihara, J. N. Kondo, E.Sato, K. Mitsudo, S. Suga, S. Iguchi, K. Kamiya, M. Atobe: J. Am. Chem. Soc. 146 (2024) 30212-30221.
[2] E. Vitaku, D. T. Smith, J. T. Njardarson: J. Med. Chem. 57 (2014) 10257-10274.
[3] P. Bhutani, G. Joshi, N. Raja, N. Bachhav, P. K. Rajanna, H. Bhutani, A. T. Paul, R. Kumar: J. Med. Chem. 64 (2021) 2339-2381.
[4] F. Chen, W. Li, B. Sahoo, C. Kreyenschulte, G. Agostini, H. Lund, K. Junge, M. Beller: Angew. Chem. Weinheim Bergstr. Ger. 130 (2018) 14696-14700.
[5] W. Qian, L. Lin, Y. Qiao, X. Zhao, Z. Xu, H. Gong, D. Li, M. Chen, R. Huang, Z. Hou: Appl. Catal. A Gen. 585 (2019) 117183.
[6] F. Martinez-Espinar, P. Blondeau, P. Nolis, B. Chaudret, C. Claver, S. Castillón, C. Godard: J. Catal. 354 (2017) 113-127.
[7] J. G. Keay: Comprehensive Organic Synthesis, Elsevier. (1991) 579-602.
[8] J. N. Kondo, S. Ge, T. Suzuki, R. Osuga, T. Matsumoto, T. Yokoi, Y. Shimizu, A. Fukazawa, N. Shida, M. Atobe: J. Phys. Chem. C Nanomater. Interfaces. 126 (2022) 19376-19385.
[9] Y. Shimizu, J. Harada, A. Fukazawa, T. Suzuki, J. N. Kondo, N. Shida, M. Atobe: ACS Energy Lett. (2023) 1010-1017.
[10] Y. Furutani, Y. Shimizu, J. Harada, Y. Muto, A. Yonezawa, S. Iguchi, N. Shida, M. Atobe: ACS Catal. 14 (2024) 8922-8929.
横浜国立大学 大学院工学研究院
〒240-8501 神奈川県横浜市保土ヶ谷区常磐台79-5
e-mail : shida-naoki-gz@ynu.ac.jp
横浜国立大学 大学院工学研究院
〒240-8501 神奈川県横浜市保土ヶ谷区常磐台79-5
e-mail : atobe-mahito-wk@ynu.ac.jp
京都大学 大学院工学研究科
〒615-8510 京都府京都市西京区京都大学桂A4棟128号室
e-mail : iguchi.shoji.4k@kyoto-u.ac.jp




