



Volume 28, No.2 Pages 113 - 118
1. 最近の研究から/FROM LATEST RESEARCH
SACLAで捉えたフラビンタンパク質中の逐次的電子・プロトン移動の動的制御
Dynamic Control of Sequential Electron and Proton Transfer Events in a Flavoenzyme Captured at SACLA
[1]大阪大学 大学院基礎工学研究科 Graduate School of Engineering Science, Osaka University、[2]Academia Sinica, Taiwan、[3]Department of Chemistry, National Taiwan University、[4]Department of Chemistry, Philipps University Marburg, Germany、[5]Institute of Biological Chemistry, Academia Sinica, Taiwan、[6](国)理化学研究所 放射光科学研究センター RIKEN SPring-8 Center、[7]東京大学 大学院農学生命科学研究科 Graduate School of Agricultural and Life Sciences, The University of Tokyo
- Abstract
- フラビン補酵素はさまざまな生体内酸化還元反応において普遍的に用いられている。フラビンアデニンジヌクレオチド(FAD)を有するDNA光回復酵素は、DNA修復反応およびFADの光還元反応に青色光を用いる。後者の反応では、FADへの2回の電子移動反応およびプロトン化が起こることで、DNA修復活性を有する酵素状態が形成される。本研究では、光還元における電子移動反応に続くナノ秒~マイクロ秒におけるFADおよび周辺アミノ酸側鎖の動きを記述するため、SACLAにて時分割シリアルフェムト秒X線結晶構造解析を実施した。その結果、光回復酵素・クリプトクロムスーパーファミリーが持つFAD近傍に存在するAsn側鎖およびArg–Asp塩橋が、FAD還元反応に伴う構造変化およびプロトン化を制御することを見出した。
1. フラビンと光回復酵素
光合成や好気呼吸に代表されるように、全ての細胞システムは電子伝達鎖に依存している[1][1] I. Belevich, D. A. Bloch, N. Belevich, M. Wilström and M. I. Verkhovsky: Proc. Natl. Acad. Sci. USA 104 (2007) 2685-2690.。シトクロムや鉄硫黄タンパク質のような電子伝達タンパク質群は一電子移動を行う一方で、電子伝達鎖が触媒する反応サイクルではドナー・アクセプター間で計2電子の移動が行われる。この一見した矛盾は、キノン類分子もしくはフラビン系分子の存在によって解決される。これらの分子種は1電子ないし2電子を受け取ることができるため、酸化型(キノン)、半還元型(セミキノン)、ならび還元型(ヒドロキノン)の3つの酸化還元状態で存在する。フラビンアデニンジヌクレオチド(FAD)における複数の電子移動反応の一例として、DNA修復を担うフラビンタンパク質であるDNA光回復酵素におけるFAD光還元反応が挙げられる。光還元反応とは、サブナノ秒で起こる光誘起電子移動を起点として、触媒不活性な酸化型FAD(FADox)からラジカルセミキノンFAD•–およびそのプロトン化体FADH•を経て、光回復酵素のDNA修復活性に必要なヒドロキノンFADH–を光依存的に生成する過程である(図1A)[2][2] C. Aubert, M. H. Vos, P. Mathis, A.P. Eker and K. Brettel: Nature 405 (2000) 586-590.。この反応の電子伝達鎖はタンパク質内部に存在する3つないし4つのTrp側鎖であり、最終的に外部環境から電子を獲得することができる[3][3] K. Brettel and M. Byrdin: Curr. Opin. Struct. Biol. 20 (2010) 693-701.。活性化された光回復酵素は、光励起状態のFADH–*から紫外線によって形成される損傷DNAであるシクロブタン型ピリミジンダイマーへ青色光依存的に電子移動が起こることで、元の塩基構造へと戻すことができる[4][4] A. Sancar: Chem. Rev. 103 (2003) 2203-2238.。
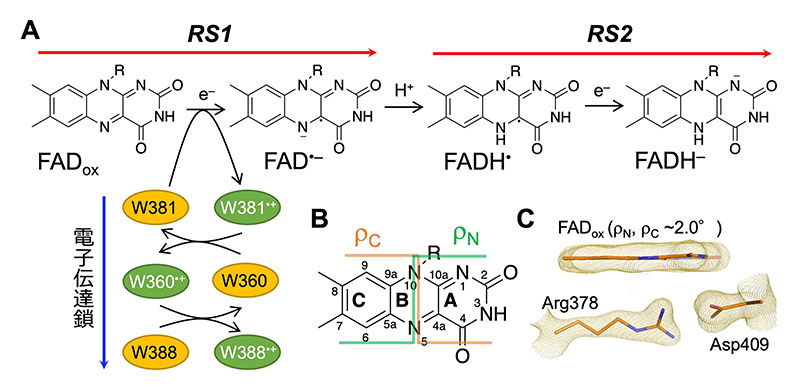
図1 光回復酵素中のFADについて。(A) FAD光活性化過程。光回復酵素中に高度に保存された3つのTrp(W)側鎖から光依存的に電子を獲得することで、酸化型FAD(FADox)からアニオンラジカル型FAD(FAD•–)へと変化し、さらにプロトン化が進行することで中性セミキノン型FAD(FADH•)が生じる。FADH•が同様の反応を経ることで、2電子還元型FAD(FADH–)が生じる。(B) FADの構造変化を記述する二面角。C9–N10–N5–C4をρC、C6–N5–N10–N1をρNと定義する。(C) FADoxを有するMmCPDIIのSFX構造と2Fo-Fc電子密度マップ。
過去30年以上にわたり、光回復酵素のFAD光還元反応は分光学や計算科学によって研究されてきた。しかし、ナノ秒~マイクロ秒に起こる電子移動後のフラビン化学種がどのように周辺アミノ酸側鎖によって安定化されるのか、依然として情報が不足している。さらに、分光学測定ではフラビン化学種や電子伝達鎖の電子遷移に限定した情報を与えるため、タンパク質環境やフラビンの構造変化に関する情報は得られない。従来のX線結晶構造解析では、短寿命なフラビンラジカル類を含めた過渡的な立体構造の捕捉は困難であり、さらに放射線損傷の影響により単一の酸化還元状態にある構造さえも解析することは難しい。
本研究では、Methanosarcina mazei由来クラスII DNA光回復酵素(MmCPDII)の光還元反応に着目し、FAD酸化還元状態の変化、および過渡的な立体構造変化をSACLAにおける時分割シリアルフェムト秒X線結晶構造解析(TR-SFX)により解析した。本研究により、フラビンラジカル類ならびに過渡生成化学種がどのように安定化されるかを明らかにした[5][5] M. Maestre-Reyna et al.: Nat. Chem. 14 (2022) 677-685.。
2. 実験概要と構造命名ルール
光回復酵素の光還元反応では、1段階目の還元反応(RS1)としてFADoxからFAD•–の形成を経てFADH•が、また2段階目の反応(RS2)ではFADH•からFADH–の形成が起こる。RS1において、FAD•–のプロトン化が起こらない限り、RS2は起こらない。そこで、FADox、FADH•ならびにFADH–を有する酵素を各々調製し、RS1とRS2を別々に追跡した。得られた酵素の構造はEX/Yと表し、Xは反応前のFAD酸化還元状態(ox:FADox、semi:FADH•、red:FADH–)を、またYはデータ収集条件(sync:シンクロトロン、ss:定常状態SFX、dark:TR-SFXにおける暗状態構造、時間:TR-SFXにおける遅延時間)とする。
また、FAD中のイソアロキサジン環の構造変化を記述するため、2つの二面角ρCおよびρNを導入する(図1B)。一般に、FADの還元に伴って、A環面とC環面間の角度αが小さくなるバタフライ構造をとることが知られている。しかし、角度αは長軸側のねじれ構造と水平方向の曲がりや短軸側のバタフライ構造を区別することができない。二面角の導入により、これらを区別することができる。
3. RS1における構造変化
RS1の初期状態であるEox/ssの無損傷SFX測定の結果、シンクロトロン構造Eox/sync [6][6] S. Kiontke et al.: EMBO J. 30 (2011) 4437-4449.と比較して、同様の全体構造をとっていることを確認した。しかし、Eox/syncにおけるイソアロキサジン二面角(ρC~7.6°, ρN~8.7°)と比較して、Eox/ssはよりフラットな構造(ρC, ρN~2.0°)を有することがわかった(図1C)。このことから、Eox/ssはほぼ完全にFADoxを持つ一方で、Eox/syncは放射線損傷を受け、部分的に還元されていることが示唆された。また、Eox/darkはEox/ssとほぼ同一の構造であったことから、TR-SFX測定のコントロールとなることがわかった。
FADoxを有する酵素結晶を408 nmにて励起後、10 nsから5 msまでの9点の遅延時間にて、RS1の電子移動後に起こる構造変化を追跡した(図2A)。その結果、イソアロキサジン環や周辺環境における構造変化が見られた一方で、電子伝達鎖であるTrpには変化が見られなかった。この結果は本酵素の電子移動そのものは1 ns以下にて完了するという分光学による知見と一致する。
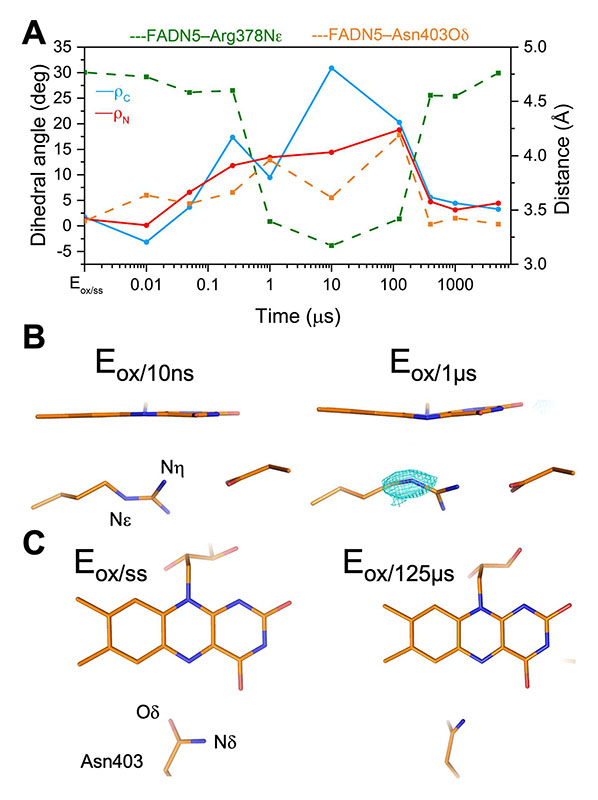
図2 RS1におけるFADおよび近傍アミノ酸の構造変化。(A) FADおよびArg378–Asp409塩橋の構造変化。青色はEox/1µs–Eox/ssの電子密度差マップ(3σ)の正の変化を表している。(B) FADおよびAsn403の構造変化。(C) RS1におけるFAD構造変化(実線:ρCとρN)および周辺アミノ酸との距離の変化(点線)。
まず、FAD構造の変化について記述する。FADoxからFAD•–の遷移に伴い、ρCが10 nsから10 µsにかけて約30°まで増加した(図2A)。ρNについても同様の傾向を示したが、その変化は緩やかであった。一方、2つの二面角は125 µsから400 µsにかけて元の角度へと戻った。これは過渡的に形成されたトリプトファニルラジカルとFAD•–の間で電荷再結合することで暗状態へと戻っていることを示唆している。
FADの近傍に、Arg378とAsp409で形成される塩橋があり、このArg378において構造変化が認められた。Eox/1µs–Eox/ssの差マップを解析したところ、Arg378のNε原子の上部に正の電子密度ピークが観測された(図2B)。このピーク強度は250 nsから1 µsにかけて増大し、その後1–5 msにかけて元の状態に緩和した[5][5] M. Maestre-Reyna et al.: Nat. Chem. 14 (2022) 677-685.。このことから、Arg378のイソアロキサジンN5原子への接近は、FAD•–の折れ曲がりと同時に起こっていることが示唆された。興味深いことに、Arg378のNεがN5へ接近する一方で、Arg378–Asp409塩橋には変化が観測されなかった。
上記N5の近傍には、Asn403が位置している。Eox/ss構造では、Asnアミド基NδはイソアロキサジンO4と水素結合距離に位置している一方で、RS1の進行に伴いアミド基が回転し、NδはイソアロキサジンN5に接近する様子が観測された(図2C)。
4. RS1後のEsemi/ss構造
RS1の後、FAD•–がプロトンを獲得すると、FADH•が形成される。RS1のTR-SFX測定では、10 µs以降暗状態への電荷再結合が観測されていたため、FADH•形成過程を捉えることはできなかった。そこで、RS2の初期状態であるEsemi/ssの無損傷SFX測定を行った。
Esemi/ssのイソアロキサジンは比較的フラット(ρC, ρN ~5°)であり、FADH•の構造はFADoxのそれと類似点が見られた(図3A)。一方で、その近傍に存在するArg378–Asp409塩橋には変化が見られた。Asp409の構造は大きな変化がなかった一方で、Arg378のグアニジン部位は回転しており、もはやArg–Asp側鎖間で塩橋は形成していないことが観測された(図3A)。この結果および周辺環境にプロトンドナーとなりうるアミノ酸側鎖や水分子が存在しないことから、Arg378がFAD•–からFADH•へのプロトンドナーを担っていることが示唆される。実際、19,524種の光回復酵素・クリプトクロムスーパーファミリータンパク質のアミノ酸配列を調査すると、わずか9種を除く大多数がこの塩橋を保有しており、高度に保存されている塩橋が機能に非常に重要であることが示唆される。
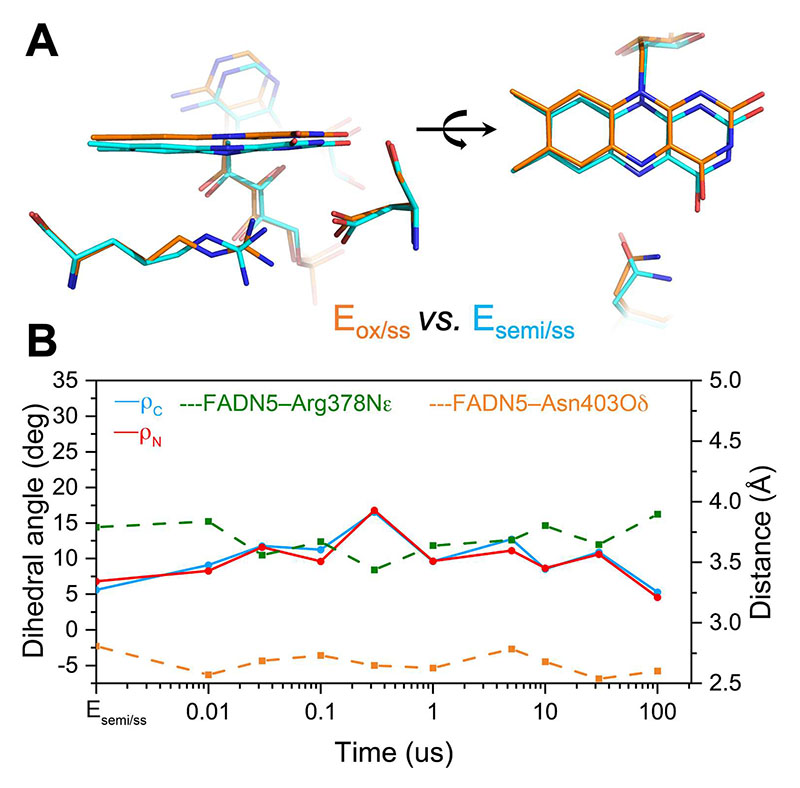
図3 RS2におけるFADおよび近傍アミノ酸の構造変化。(A) RS2の初期状態であるEsemi/ssの構造(シアン)。比較のため、RS1の初期状態であるEox/ssの構造(オレンジ)についても示している。(B) RS2におけるFAD構造変化(実線:ρCとρN)および周辺アミノ酸との距離の変化(点線)。
溶液中におけるFADH•のpKaは8.5程度であることから、塩基性側鎖(pKa ~12.5)を持つArgからのプロトン化は起こらないことが考えられる。しかし、フラボドキシン中のFMNH•のpKaは、タンパク質主鎖のカルボニル基との水素結合形成により、13程度まで上昇することが報告されている [7][7] M. L. Ludwig, L. M. Schopfer, A. L. Metzger, K. A. Pattridge and V. Massey: Biochemistry 29 (1990) 10364-10375.。今回、同様のことがMmCPDIIでも起こっていると考えられる。Eox/ssにおけるAsn403のOδ原子は、イソアロキサジンN5原子の近傍に位置しているが、FAD•–の形成に伴い回転し、N5から遠ざかる様子が観測されていた(図2C)。プロトン化が起こった後のEsemi/ssでは、Asn403側鎖は再び回転し、N5–Hの近傍に位置していた(図3A)。このことから、Asn403がFADH•のpKaの向上に寄与すると考えられる。
5. RS2における構造変化
次に、FADH•からFADH–への光還元反応(RS2)の追跡のため、FADH•を有する酵素微結晶を408 nmで励起後、10 nsから100 µsまでの9点の遅延時間にて構造解析を行なった。二面角ρCとρNは300 nsまで一様に増加し、16.5°および16.8°まで変化した(図3B)。この値は、Ered/ssにおける二面角の値(14.3°と14.5°)とよい相関を示す。しかし、これらの値は100 µsにかけて5°程度戻ることから、部分的に電荷再結合が進行していることが示唆された。
RS2では、イソアロキサジン二面角が最も大きくなる300 nsまで、イソアロキサジンN5とArg378のNε原子間距離が短くなる様子が観測された。一方で、Arg378側鎖の位置そのものはRS1での観測と同様に、大きな変化は観測されなかった。つまり、この接近はイソアロキサジン環の折れ曲がりに由来するものであると考えられる。Asp409の位置もEsemi/ssから大きな変化はみられなかった。また、Asn403のOδはイソアロキサジンN5–Hからやや遠位にシフトしたものの、RS2の過程で大きく変化していなかった。
6. 光回復酵素の光還元反応機構
今回のTR-SFX測定で捉えたRS1とRS2におけるナノ秒からマイクロ秒で起こる構造変化、ならびFADの各酸化還元状態のSFX構造から、光還元反応の全反応機構について総括する(図4)。
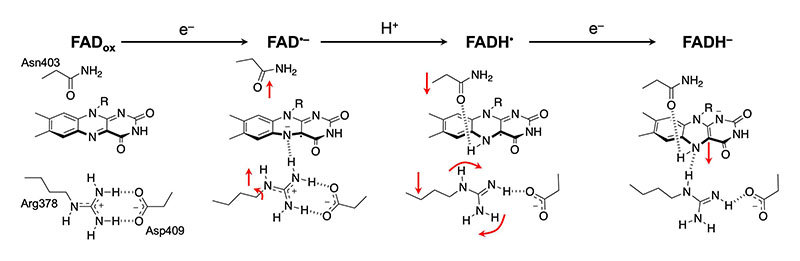
図4 TR-SFX測定で捉えたFADおよび酸化還元センサーであるArg–Asp塩橋ならびAsnの構造変化。
光励起後のFADoxは電子伝達鎖のTrp側鎖から電子を獲得し、FAD•–が過渡的に形成する。FADの電子状態の変化に伴って、Asn403のOδは静電反発により回転しイソアロキサジンN5から離れる一方で、Asn403のNδおよび正電荷を持つArg378側鎖が静電相互作用によって引き付けられる。結果、1 µsから125 µsの間でイソアロキサジンN5とArg378のNη間の距離が最も短く、またAsn403のOδとの距離が最も離れ、イソアロキサジンN5のプロトン化に備える。
Arg378側鎖がイソアロキサジンN5に最も近づいた後、プロトン移動が起こる。これにより生成するFADH•およびArg378は中性電荷であり、相互作用の弱体化に伴ってArg378はイソアロキサジンおよびAsp409から離れる。この状態では、Arg378–Asp409はもはや塩橋ではなく、1本の水素結合で結ばれている。プロトン移動の駆動力はAsn403のOδとN5–Hとの水素結合であり、これが中性セミキノン状態における主要な安定化要因となる。
RS2では、Asn403のOδがイソアロキサジンからやや遠ざかるものの、N5–Hとの水素結合を維持しつつ、100 nsから1 µsにかけて起こるイソアロキサジンの折れ曲がりによって、N5原子がArg378のNεへと接近する。これによって、還元状態FADH–の構造とほぼ同様の状態になる。この際、上記2種類の水素結合がFADH–の安定化に寄与するものと考えられる。
これらのことから、イソアロキサジン近傍に存在するAsnおよびArg–Asp塩橋は、FADの酸化還元センサーとして機能し、プロトン化で分断されたキノン類の2電子還元反応を制御していることがわかった。
7. 総括と今後の展開
本研究で見つかったAsn/Arg–Aspセンサーについて、Arg–Asp(Glu)対をイソアロキサジン環近傍に持つフラビンタンパク質群を構造ベースで探索したところ、光回復酵素・クリプトクロムスーパーファミリーのみが持つことがわかり[5][5] M. Maestre-Reyna et al.: Nat. Chem. 14 (2022) 677-685.、本タンパク質ファミリーの特徴的な役割を果たすことが示唆された。興味深いことに、Asnについては酵素活性を持つ光回復酵素にのみ保存されていることが知られている。植物由来の光受容クリプトクロムではAspを、また昆虫型クリプトクロムではCysをAsnの代わりに持ち、それぞれ光還元反応においてFADH•およびFAD•–状態を安定化することが知られている[8][8] I. Chaves et al.: Annu. Rev. Plant Biol. 62 (2011) 335-364.。つまり、オルタナティブな酸化還元センサーによってフラビンの酸化還元反応が大幅に制御されることからも、本センサーの重要性を窺い知ることができる。
光回復酵素の分子内電子移動反応は、光合成反応中心における光駆動電荷分離過程と同等の最も単純なモデル系であると認識されてきた。QBキノン電子受容体での電子供給は、電子移動–プロトン移動–電子移動–プロトン移動のスキームを取ることが知られており、酸化還元状態に依存したQB分子の移動を伴う。今回、Arg側鎖がフラビンのプロトン化に関与するという予期せぬ観測結果から、今後光合成反応中心のような複雑生命系における酸化還元反応のTR-SFX測定を行うことで、これらの中で起こる酸化還元反応に伴うプロトン化過程を明らかにできることが期待される。
謝辞
本研究は、SACLAのBL2(課題番号2017A8019、2017B8052、2018A8008、2018B8031、2019A8014、2019B8005)にて実施されました。本研究の一部は科研費(16K01942、21K06042)の助成を受けて行われました。
参考文献
[1] I. Belevich, D. A. Bloch, N. Belevich, M. Wilström and M. I. Verkhovsky: Proc. Natl. Acad. Sci. USA 104 (2007) 2685-2690.
[2] C. Aubert, M. H. Vos, P. Mathis, A.P. Eker and K. Brettel: Nature 405 (2000) 586-590.
[3] K. Brettel and M. Byrdin: Curr. Opin. Struct. Biol. 20 (2010) 693-701.
[4] A. Sancar: Chem. Rev. 103 (2003) 2203-2238.
[5] M. Maestre-Reyna et al.: Nat. Chem. 14 (2022) 677-685.
[6] S. Kiontke et al.: EMBO J. 30 (2011) 4437-4449.
[7] M. L. Ludwig, L. M. Schopfer, A. L. Metzger, K. A. Pattridge and V. Massey: Biochemistry 29 (1990) 10364-10375.
[8] I. Chaves et al.: Annu. Rev. Plant Biol. 62 (2011) 335-364.
大阪大学 大学院基礎工学研究科
〒560-8531 大阪府豊中市待兼山町1-3
TEL : 06-6850-6240
e-mail : yamamoto.junpei.es@osaka-u.ac.jp
Academia Sinica, Taiwan
Department of Chemistry, National Taiwan University
e-mail : mmaestre@ntu.edu.tw
Department of Chemistry, Philipps University Marburg, Germany
e-mail : essen@chemie.uni-marburg.de
Institute of Biological Chemistry, Academia Sinica, Taiwan
e-mail : mdtsai@gate.sinica.edu.tw
Academia Sinica, Taiwan
(国)理化学研究所 放射光科学研究センター
東京大学 大学院農学生命科学研究科
〒113-8657東京都文京区弥生1-1-1
TEL : 03-5841-3069
e-mail : bessho@g.ecc.u-tokyo.ac.jp




