



Volume 25, No.3 Pages 211 - 216
1. 最近の研究から/FROM LATEST RESEARCH
DXAFS法を利用した自動車触媒用酸素貯蔵材料の動的挙動観察
Observation of Dynamic Behaviour of Oxygen Storage Material for Automotive Catalysts using DXAFS
[1]京都大学 触媒・電池元素戦略研究拠点 Elements Strategy Initiative for Catalysts & Batteries (ESICB), Kyoto University、[2]龍谷大学 先端理工学部 Department of Materials Chemistry, Ryukoku University、[3](公財)高輝度光科学研究センター 放射光利用研究基盤センター 分光・イメージング推進室 Spectroscopy and Imaging Division, Center for Synchrotron Radiation Research, JASRI、[4]京都大学 大学院工学研究科 Graduate School of Engineering, Kyoto University
- Abstract
- 自動車触媒の必要構成要素である酸素貯蔵材料の開発を行っており、最近、Sr3Fe2O7-δが優れた酸素貯蔵能を示すことを見出した。酸素貯蔵速度の評価は、系内に水素もしくは酸素ガスを瞬時に導入した際の酸素放出・貯蔵挙動をin-situ DXAFS法により測定することで行った。その結果、Sr3Fe2O7-δにPdを担持することで、反応律速であった表面での水素乖離が促進し、劇的に酸素放出速度が向上することを精度よく分析することに成功した。また、異種元素置換型Sr3Fe2O7-δにおいて、Feサイトを一部Mnに置き換えた材料が、高い酸素放出速度を示すことも見出した。Mn置換材料の結果から、酸素放出には水素乖離などの表面反応だけでなく、酸素放出前後の微小な構造変化も大きな影響を与えることを明らかにした。
1. はじめに
ガソリンエンジンを搭載した自動車の排ガス中には、有害物質であるNOxやCO、未燃焼の炭化水素が含まれている。これら有害物質は自動車触媒によってCO2やN2、H2Oに分解され大気中に放出されている。現行の自動車触媒は、主にPdおよびRhを含んだ貴金属触媒とCeO2系酸素貯蔵材料で構成されており[1,2][1] J. Kašper, P. Fornasiero and N. Hickey: Catal. Today 77 (2003) 419-449.
[2] R. J. Farrauto, M. Deeba and S. Alerasool: Nat. Catal. 2 (2019) 603-613.、厳しい排ガス規制を満たし、高い浄化性能を有するために多量の貴金属が使用されている。そのため、元素戦略の観点から貴金属使用量の低減化および貴金属代替が期待されているとともに、抜本的な機能向上も求められている。
貴金属触媒の欠点は、酸素濃度が変動した場合に浄化効率は大きく低下するところにある。ところが、実際の排ガス中の酸素濃度は、走行モードに応じて常に変動しているため、周囲の雰囲気に応じて酸素を可逆的に吸蔵・放出することのできる酸素貯蔵材料が自動車触媒には必要不可欠な要素となっている。
現在実用化されている酸素貯蔵材料はCeO2-ZrO2固溶体であり、酸素吸蔵および放出過程における構造変化では、CeおよびZrサイトの配列を維持した状態で酸化物イオンのみが挿入・脱離している[3-5][3] M. Sugiura, M. Ozawa, A. Suda, T. Suzuki and T. Kanazawa: Bull. Chem. Soc. Jpn. 78 (2005) 752-767.
[4] Y. Nagai, T. Yamamoto, T. Tanaka, S. Yoshida, T. Nonaka, T. Okamoto, A. Suda and M. Sugiura: Catal. Today 74 (2002) 225-234.
[5] T. Yamamoto, A. Suzuki, Y. Nagai, T. Tanabe, F. Dong, Y. Inada, M. Nomura, M. Tada and Y. Iwasawa: Angew. Chem. Int. Ed. 46 (2007) 9253-9256.。このように結晶学的な関係がみられる構造相転移はトポタクティック転移とも呼ばれている。すなわち、CeO2-ZrO2固溶体の酸化物イオンの振る舞いは、リチウムイオン電池の正極材料であるコバルト酸化物へのリチウムイオンの挿入脱離機構に類似している。
最近、我々はトポタクティックな酸化物イオンの挿入・脱離機構に着目し、新規酸素貯蔵材料の設計を行っている。その中でも、層状ペロブスカイト構造を持つSr3Fe2O7-δはCeO2-ZrO2固溶体と同程度の優れた酸素貯蔵特性を示すことを明らかにした[6][6] K. Beppu, S. Hosokawa, K. Teramura and T. Tanaka: J. Mater. Chem. A 3 (2015) 13540-13545.。また、本材料は自動車排ガス浄化反応に対する触媒担体としても有効に作用することを見出している[7][7] K. Beppu, S. Hosokawa, H. Asakura, K. Teramura and T. Tanaka: Catal. Sci. Technol. 8 (2018) 147-153.。
本稿では、Sr3Fe2O7-δの酸素貯蔵能に対するPd担持効果および異種元素置換効果をその場波長分散型X線吸収分光法(in-situ DXAFS)で評価した実例を紹介する。
2. in-situ DXAFS測定の概要
実験室におけるSr3Fe2O7-δの酸素貯蔵能測定は、熱重量分析(TG)装置を用いて行っている。例えば、酸素貯蔵量は、反応温度を所定温度に固定し、水素ガスおよび酸素ガスを交互に切り替えた際の重量変化量から見積もっている。TG装置においても、基質ガス(H2もしくはO2)を導入した直後の重量変化速度から、酸素貯蔵速度および放出速度を見積もることはできる。ところが、系内のガス置換速度を無視することはできず、測定における時間分解能も高くないという問題点がある。そこで、ミリ秒オーダーの時間分解能を持つ波長分散型X線吸収分光法(DXAFS)を利用することで、酸素吸蔵および放出速度を精度よく分析することを試みた。
本装置の概略図を図1に示す。DXAFS法に使用するバッチセルは、ガス流通および真空に引くことができるように設置し、実際の測定は予め真空に引いたセル内に1気圧の水素ガスもしくは酸素ガスを導入することで行った。そのため、ガスの拡散はほぼ無視することができる。
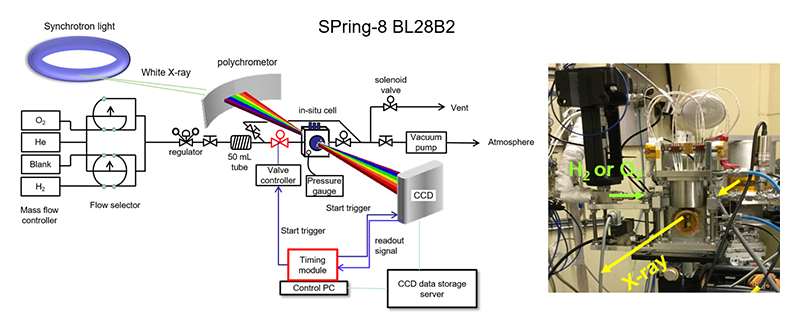
図1 in-situ DXAFS実験装置の概略図。
Sr3Fe2O7-δの酸素放出・貯蔵過程では、式(1)および(2)で示すようにFe種のレドックスが作用している。
| Sr3Fe2O7-δ + (1-δ)H2 → Sr3Fe2O6 + (1-δ)H2O | ・・・ (1) |
| Sr3Fe2O6 + 1/2(1-δ)O2 → Sr3Fe2O7-δ | ・・・ (2) |
そのため、通常はFe K-edge X線吸収端近傍構造(XANES)のedge位置を測定することを想定するが、あいにくFe4+とFe3+のedge位置の変化量は少ない。一方、Sr3Fe2O7-δのSr K-edge XANESスペクトルのポストエッジの高さは、酸素放出前後で大きく変化する(図2)。この実験事実を基に、Sr K-edgeをDXAFS測定の対象とした。なお、本測定では、試料設置部を550−600°Cに加熱し、スペクトルを約70 ms毎に測定した。

図2 Sr3Fe2O7-δのSr K-edge XANESスペクトル。(黒線)酸素放出前の試料、(赤線)酸素放出後の試料。
3. Pd/Sr3Fe2O7-δの酸素貯蔵能[8][8] K. Beppu, S. Hosokawa, T. Shibano, A. Demizu, K. Kato, K. Wada, H. Asakura, K. Teramura and T. Tanaka: Phys. Chem. Chem. Phys 19 (2017) 14107-14113.
図3(A)に、作動温度500°CにおけるSr3Fe2O7-δのTG測定結果を示す。Pdの担持の有無による酸素貯蔵量および酸素放出速度の変化は、本測定において有意な差は見られなかった。そこで、in-situ DXAFS測定によりSr3Fe2O7-δの酸素放出速度を評価した(図3(B))。図3(B)における縦軸は水素導入時に連続して変化したスペクトルを酸素放出前後のスペクトルで線形結合近似することで見積もった酸素放出挙動である。そのため、酸素放出前のスペクトルを1とし、酸素放出後のものを0として規格化している。その結果、Sr3Fe2O7-δは水素導入後、緩やかに格子酸素が放出されて行き、約80秒後に式(1)の反応が完了した。ところが、Pd/Sr3Fe2O7-δでは、酸素放出速度が劇的に向上し、およそ300 ms後にはPd/Sr3Fe2O7-δからPd/Sr3Fe2O6への格子酸素の放出が完了していた。また、Pd/Sr3Fe2O7-δでは水素導入直後にフラクションが負の値となっている。これは層状ペロブスカイト構造に起因する中間相の形成を捉えているものと現在考えている。なお、酸素貯蔵速度についても同様の手法で検討したが、Pdの担持効果は顕著に現れなかった。
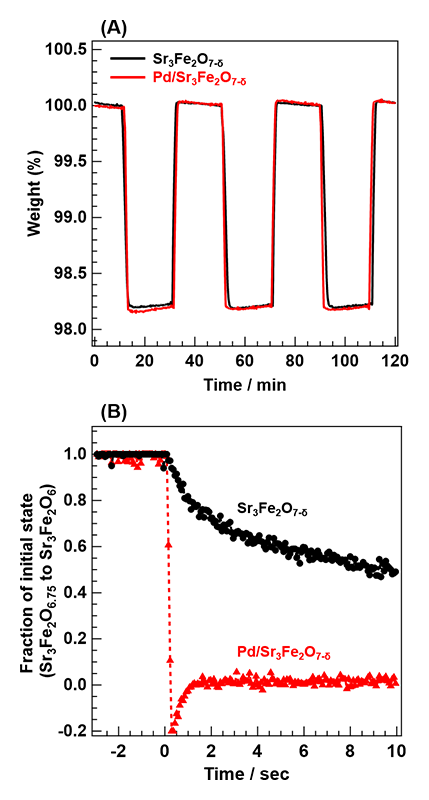
図3 (A) TG測定におけるSr3Fe2O7-δおよびPd/Sr3Fe2O7-δの酸素吸蔵・放出挙動。(B) in-situ DXAFS測定におけるSr3Fe2O7-δおよびPd/Sr3Fe2O7-δの酸素放出挙動。
水素雰囲気下での昇温反応を行ったところ、Pdの担持により、格子酸素放出温度が500°Cから130°Cへ大きく低温化していることも認められた。一方、不活性ガス(He)雰囲気下での格子酸素放出温度は、Pd担持により低温化していなかった。これらの結果から、Sr3Fe2O7-δ上でのH2の乖離が酸素放出の律速段階であり、それをPdが促進しているため、酸素放出速度の向上および酸素放出温度の低温化が実現したものと結論した。
4. 異種元素置換型Sr3Fe2O7-δの酸素貯蔵能[9][9] K. Beppu, S. Hosokawa, A. Demizu, Y. Oshino, K. Tamai, K. Kato, K. Wada, H. Asakura, K. Teramura and T. Tanaka: J. Phys. Chem. C 122 (2018) 11186-11193.
Feサイトに異種元素遷移金属を置換させたSr3(Fe0.8M0.2)2O7-δ(M = Ni, Mn)の酸素貯蔵量をTG装置にて評価した(図4(A))。その結果、Mnを固溶させた試料の酸素貯蔵量は、Sr3Fe2O7-δ(1.9 wt%)と同程度であった。ところが、Niを固溶させた試料(2.2 wt%)は酸素貯蔵量の向上が認められた。次に、酸素放出・貯蔵速度をin-situ DXAFS法により評価した。図4(B)に記載している縦軸は、TG装置にて求めた酸素貯蔵量により規格化している。Sr3Fe2O7-δの酸素放出速度は、特にMnを置換した試料において大幅に向上した。

図4 (A) TG測定におけるSr3(Fe0.8M0.2)2O7-δの酸素吸蔵・放出挙動。(B) in-situ DXAFS測定におけるSr3(Fe0.8M0.2)2O7-δの酸素放出挙動。
そこで、Sr3(Fe0.8M0.2)2O7-δの酸素放出前後の遷移金属サイトの局所構造を静的なXAFS測定により評価した。酸素放出前のSr3(Fe0.8Ni0.2)2O7-δにおいて、Fe K-edgeおよびNi K-edgeのEXAFS振動はk = 12付近まで明瞭に観測された(図5)。ところが、酸素放出後、それらのEXAFS振動の強度は低下し、振動パターンが大きく変化した。また、Ni K-edge XANESのedge位置は、酸素放出に伴って低エネルギー側に大きく移動しており、概ねNi2+まで還元されていた。粉末X線回折(XRD)においてNiOのような不純物が検出されなかったことから、Sr3(Fe0.8Ni0.2)2O7-δ結晶内の遷移金属種周りの局所構造の秩序は酸素放出に伴って大きく乱れたことが予想できる。また、酸素貯蔵時におけるSr3Fe2O7-δ中のFe種はFe4+とFe3+の混合状態であり、その中のFe4+種のみが酸素放出に寄与している。一方、酸素放出後のSr3(Fe0.8Ni0.2)2O7-δではFe4+の還元に加え、Ni3+種がNi2+まで還元されたものと考えられる。なお、Ni3+種はSr3Fe2O7-δの酸素放出に関与していないFe3+サイトに選択的に固溶していると考察しており、その固溶状態とNi種特有のレドックス機構によりSr3(Fe0.8Ni0.2)2O7-δはSr3Fe2O7-δより優れた酸素貯蔵量を示したものと結論した。
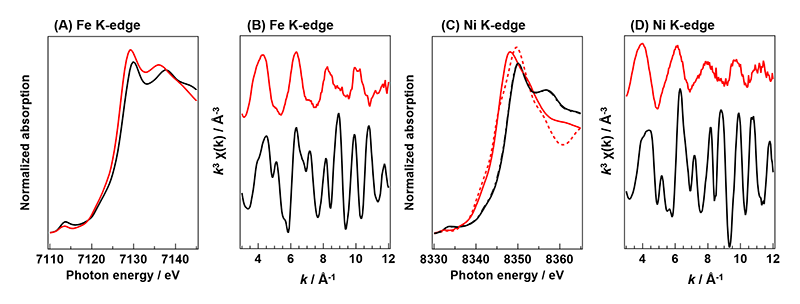
図5 Sr3(Fe0.8Ni0.2)2O7-δのFe K-edgeおよびNi K-edge XAFSスペクトル。(A),(C) XANESスペクトル、(B),(D) EXAFS振動。(黒線)合成直後の試料、(赤線)酸素放出後の試料、(赤点線)NiO。
一方、Sr3(Fe0.8Mn0.2)2O7-δのFe K-edgeおよびMn K-edgeのEXAFS振動は、酸素放出後においてもk = 12付近まで明瞭に観測された(図6)。特に、酸素放出後の試料の振動パターンは合成直後のものと類似していた。この結果は、酸素放出後も合成直後の局所構造を概ね維持していることを示している。さらに、XRD測定より見積もった酸素放出前後の単位格子体積変化は、Niに比べてMnを固溶させたものが小さかった。また、水素流通下での昇温反応により格子酸素放出温度を評価したところ、Pd担持のような劇的な低温化は起きていなかった。このことから、Sr3(Fe0.8Mn0.2)2O7-δは酸素放出前後の構造変化が極めて小さいため、酸素放出速度が向上したものと考えている。なお、Mn K-edgeのXANESスペクトルの酸素放出に伴うピークシフトは顕著に見られなかったことから、Mn種はNi種のようにMn2+まで還元されていない。そのため、Mn置換による酸素貯蔵量の向上は僅かであったものと考えられる。
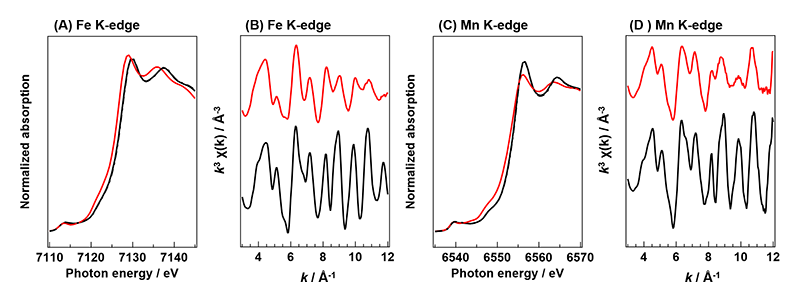
図6 Sr3(Fe0.8Mn0.2)2O7-δのFe K-edgeおよびMn K-edge XAFSスペクトル。(A),(C) XANESスペクトル、(B),(D) EXAFS振動。(黒線)合成直後の試料、(赤線)酸素放出後の試料。
以上の結果から、Sr3Fe2O7-δの遷移金属サイトのレドックス状態は添加元素により制御することが可能であり、Ni添加試料のように遷移金属サイトの価数変動が大きな場合において高い酸素貯蔵量を示すことを見出した。また、酸素放出速度は、酸素放出前後の構造変化が影響を及ぼしており、遷移金属サイトの局所構造が大きく変化しないことが好ましいことを明らかにした。最近では、本評価法を応用することで、排ガス成分であるNOとSr-Fe系複合酸化物の格子酸素の反応性についても検討することができるようになり、固体内の酸化物イオンの移動能がNO酸化反応に強く影響を及ぼしていることも見出している[10][10] K. Tamai, S. Hosokawa, K. Onishi, C. Watanabe, K. Kato, H. Asakura, K. Teramura and T. Tanaka: ACS Catal. 10 (2020) 2528-2537.。
5. Pd/Sr3(Fe0.8M0.2)2O7-δの自動車排ガス浄化特性[11][11] K. Beppu, S. Hosokawa, H. Asakura, K. Teramura and T. Tanaka: J. Mater. Chem. A 7 (2019) 1013-1021.
Pd担持および異種元素置換により酸素貯蔵能を向上させた材料の自動車排ガス浄化性能を評価した。自動車触媒反応に対するSr3Fe2O7-δの酸素貯蔵能の働きは、反応温度を500°Cに固定し、酸素濃度のみを変化させた際のNO浄化率から評価した(図7)。理論空燃比として想定している式(3)が成立する際の酸素濃度を基準とし、λという指標を以下のように定義した。
| 4CO + 4NO + C3H6 + 9/2O2 → 2N2 + 7CO2 + 3H2O | ・・・ (3) |
| (C3H6; 250 ppm, CO; 1000 ppm, NO; 1000 ppm, O2; 1125 ppm, He balance) | |
| λ = ([CO]a + [NO]a + [O2]a × 2) / ([CO]s + [NO]s + [O2]s × 2) | ・・・ (4) |
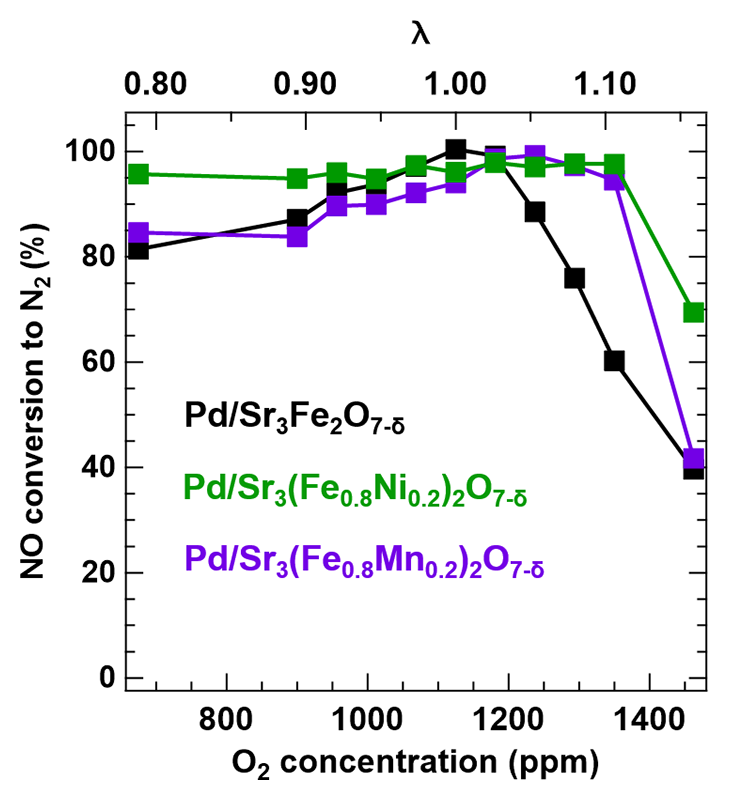
図7 Pd/Sr3Fe2O7-δ、Pd/Sr3(Fe0.8Ni0.2)2O7-δおよびPd/Sr3(Fe0.8Mn0.2)2O7-δによるNO還元活性。
なお、式(4)における[X]sは化学量論時の濃度であり、[X]aは系中に含まれる実ガス濃度を示している。すなわち、λ > 1の時は酸化雰囲気であり、λ < 1の時は還元雰囲気である。図7は、還元雰囲気(λ < 1)を経験した触媒を酸化雰囲気に移行させた際のNO還元活性を示している。Pd/Sr3Fe2O7-δ触媒はλ = 1付近まで高いNO還元活性を示した。一方、NiおよびMnを固溶させたPd/Sr3(Fe0.8M0.2)2O7-δ触媒は、Pd/Sr3Fe2O7-δ触媒よりも幅広い酸素濃度域において高い活性を示すことを見出した。特に、酸化的雰囲気下にて高い活性を示したのは、酸素貯蔵能の向上が大きく関与しているものと考えられる。
6. まとめ
ミリ秒オーダーの時間分解能を持つDXAFS法を利用することで、材料自身が持つ真の機能を精度よく評価できた。このような基礎的なデータの蓄積は、新材料の設計指針の根幹にあると思っている。さらに、酸素貯蔵能が向上した材料を自動車排ガス浄化反応に適用することで、優れた触媒活性を示すことも明らかにした。ところが、現状、触媒反応に対して有効に作用する機能(酸素貯蔵量や速度)を明確にできていないのも事実である。そのため、今後は実験室での触媒反応を完全に再現可能なOperando XAFS装置[12][12] H. Asakura, S. Hosokawa, T. Ina, K. Kato, K. Nitta, K. Uera, T. Uruga, H. Miura, T. Shishido, J. Ohyama, A. Satsuma, K. Sato, A. Yamamoto, S. Hinokuma, H. Yoshida, M. Machida, S. Yamazoe, T. Tsukuda, K. Teramura and T. Tanaka: J. Am. Chem. Soc. 140 (2018) 176-184.を積極的に活用することで、各種金属種のレドックス機構が表面反応に与える影響を詳細に検討する予定である。
参考文献
[1] J. Kašper, P. Fornasiero and N. Hickey: Catal. Today 77 (2003) 419-449.
[2] R. J. Farrauto, M. Deeba and S. Alerasool: Nat. Catal. 2 (2019) 603-613.
[3] M. Sugiura, M. Ozawa, A. Suda, T. Suzuki and T. Kanazawa: Bull. Chem. Soc. Jpn. 78 (2005) 752-767.
[4] Y. Nagai, T. Yamamoto, T. Tanaka, S. Yoshida, T. Nonaka, T. Okamoto, A. Suda and M. Sugiura: Catal. Today 74 (2002) 225-234.
[5] T. Yamamoto, A. Suzuki, Y. Nagai, T. Tanabe, F. Dong, Y. Inada, M. Nomura, M. Tada and Y. Iwasawa: Angew. Chem. Int. Ed. 46 (2007) 9253-9256.
[6] K. Beppu, S. Hosokawa, K. Teramura and T. Tanaka: J. Mater. Chem. A 3 (2015) 13540-13545.
[7] K. Beppu, S. Hosokawa, H. Asakura, K. Teramura and T. Tanaka: Catal. Sci. Technol. 8 (2018) 147-153.
[8] K. Beppu, S. Hosokawa, T. Shibano, A. Demizu, K. Kato, K. Wada, H. Asakura, K. Teramura and T. Tanaka: Phys. Chem. Chem. Phys 19 (2017) 14107-14113.
[9] K. Beppu, S. Hosokawa, A. Demizu, Y. Oshino, K. Tamai, K. Kato, K. Wada, H. Asakura, K. Teramura and T. Tanaka: J. Phys. Chem. C 122 (2018) 11186-11193.
[10] K. Tamai, S. Hosokawa, K. Onishi, C. Watanabe, K. Kato, H. Asakura, K. Teramura and T. Tanaka: ACS Catal. 10 (2020) 2528-2537.
[11] K. Beppu, S. Hosokawa, H. Asakura, K. Teramura and T. Tanaka: J. Mater. Chem. A 7 (2019) 1013-1021.
[12] H. Asakura, S. Hosokawa, T. Ina, K. Kato, K. Nitta, K. Uera, T. Uruga, H. Miura, T. Shishido, J. Ohyama, A. Satsuma, K. Sato, A. Yamamoto, S. Hinokuma, H. Yoshida, M. Machida, S. Yamazoe, T. Tsukuda, K. Teramura and T. Tanaka: J. Am. Chem. Soc. 140 (2018) 176-184.
京都大学 触媒・電池元素戦略研究拠点
〒615-8245 京都府京都市西京区御陵大原1-10
京都大学桂イノベーションプラザ
TEL : 075-383-2835
e-mail : hosokawa@scl.kyoto-u.ac.jp
龍谷大学 先端理工学部
〒520-2194 滋賀県大津市瀬田大江横谷1-5
TEL : 077-543-7466
e-mail : beppu@rins.ryukoku.ac.jp
(公財)高輝度光科学研究センター
放射光利用研究基盤センター 分光・イメージング推進室
〒679-5198 兵庫県佐用郡佐用町光都1-1-1
TEL : 0791-58-0802 ext 3949
e-mail : kkato@spring8.or.jp
京都大学 大学院工学研究科
〒615-8510 京都府京都市西京区京都大学桂
TEL : 075-383-2558
e-mail : tanakat@moleng.kyoto-u.ac.jp




