



Volume 22, No.2 Pages 110 - 115
1. 最近の研究から/FROM LATEST RESEARCH
Long-term Proposal Report 2
NRVS of Mononuclear and Binuclear Non-heme Iron Enzyme Intermediates and Related Model Complexes
[1]Department of Chemistry, Stanford University, [2]SLAC National Accelerator Laboratory, [3]Korea Advanced Institute of Science and Technology (KAIST)
- Abstract
- Mononuclear and binuclear non-heme iron enzymes catalyze a broad range of reactions that are relevant in fields from health to bioremediation. In many classes, O2 intermediates have been trapped, and knowing the structure of these intermediates is essential to understanding reactivity. We have developed a combined NRVS/DFT methodology for structural definition of these important intermediates. We present an overview of our recent work at SPring-8 applying this NRVS/DFT methodology to mononuclear FeIV-oxo and FeIII-(hydro)peroxo intermediates, and to a binuclear FeIII-peroxy P' intermediate. This has allowed us to define the structures of these intermediates and has given new insight into their reactivities.
Introduction
Non-heme iron enzymes are a diverse class of biological catalysts that drive reactions with molecular oxygen relevant in bioremediation, hypoxia regulation, DNA repair, anticancer activity, antibiotic and natural product biosynthesis. Broadly, understanding the chemistry of these systems at a fundamental level would provide great inspiration in numerous fields including drug design and bio-inspired industrial catalysts. These enzymes perform an extensive range of molecular transformations including hydrogen atom abstraction (HAA), hydroxylation, mono- and di-oxygenation, electrophilic aromatic substitution (EAS), ring cleavage/closure/expansion and halogenation reactions[1][1] Solomon, E. I.; Brunold, T. C.; Davis, M. I.; Kemsley, J. N.; Lee, S. K.; Lehnert, N.; Neese, F.; Skulan, A. J.; Yang, Y. S. and Zhou, J.: Chem. Rev. 2000, 100, 235-350.. Mononuclear non-heme enzymes (NHFe) are divided into classes based on the nature of the substrate and whether a cofactor is required (Figure 1, left)[2][2] Solomon, E. I.; Goudarzi, S. and Sutherlin, K. D.: Biochem. 2016, 55, 6363-6374.. This article summarizes studies on enzymes from two of these classes. One, the alpha-ketoglutarate (α-KG) dependent enzymes, employ an organic cofactor with a ferrous center to activate O2 by a four-electron reduction, generating an FeIV-oxo intermediate. Another class, the Rieske dioxygenases (RDOs), use a non-heme ferrous center and a reduced 2Fe2S Rieske center to reduce O2 by two electrons, potentially yielding an FeIII-(hydro)peroxy intermediate species. Binuclear non-heme iron enzymes (NH2Fe) can be classified based on whether they activate O2 by two electrons to generate biferric peroxy intermediates, P/P', or four electrons to generate a high-valent species, Q/X (Figure 1, right)[3][3] Solomon, E. I. and Park, K.: JBIC 2016, 21, 575-588.. In this review, we summarize our very recent study on P vs P' peroxide activity by the NH2Fe enzyme p-aminobenzoate N-oxygenase (AurF). Ultimately, to understand the molecular mechanism the geometric and electronic structure of these intermediates must be determined, and nuclear resonance vibrational spectroscopy (NRVS) is an ideal technique to accomplish this. NRVS is a synchrotron technique that looks at vibrational sidebands of the 57Fe Mössbauer nuclear transition[4,5][4] Seto, M.; Yoda, Y.; Kikuta, S.; Zhang, X. W. and Ando, M.: Phys. Rev. Lett. 1995, 74, 3828-3831.
[5] Sturhahn, W.; Toellner, T. S.; Alp, E. E.; Zhang, X.; Ando, M.; Yoda, Y.; Kikuta, S.; Seto, M.; Kimball, C. W. and Dabrowski, B.: Phys. Rev. Lett. 1995, 74, 3832-3835.. All normal modes with Fe displacement appear in the NRVS spectrum, with an intensity proportional to the amount of displacement at the energy of the vibrational mode, giving direct structural information on the iron active site.
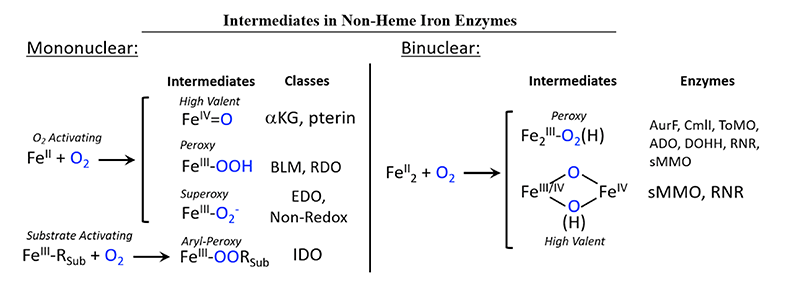
Figure 1 Mononuclear (left) and binuclear (right) non-heme Fe enzymes and their intermediates. Abbreviations: aKG - alpha-ketoglutarate dependent dioxygenase, pterin - pterin dependent hydroxylase, BLM - bleomycin, RDO - Rieske dioxygenase, EDO - extradiol dioxygenase, Non-Redox - non-redox substrate enzymes, IDO - intradiol dioxygenase, AurF - p-aminobenzoate N-oxygenase, CmlI - arylamine oxygenase of the chloramphenicol pathway, ToMO - toluene/o-xylene monooxygenase, ADO - aldehyde-deformylating oxygenase, DOHH - deoxyhypusine hydrolyase, RNR - ribonuclutide reductase, sMMO - soluble methane monooxygenase.
Methodology
Experimental details
NRVS data were collected at beamline BL09XU in C-mode. The resulting data files were analyzed with the Phoenix program package from W. Stuhrhahn[6][6] Sturhahn, W.: Hyperfine Interactions 2000, 125, 149-172.. Density functional theory (DFT) calculations were performed with Gaussian and ORCA software packages using an in-house 1,000 core cluster, with basis set and functional chosen based on model calibration (vide infra). Computational model geometries were generated either from crystal structures or EXAFS data, and modified to evaluate specific structural candidates.
DFT model calibration
We have developed a NRVS/DFT approach for defining unknown intermediate structures. This first involves collecting NRVS data on related model complexes with known structural parameters (for example, from a crystal structure or from EXAFS data). DFT calculations with a variety of functionals and basis sets are used to calculate the model complex and simulate the NRVS data, and the functional/basis set combination that best simulates the NRVS data, and any other available data, including resonance Raman and Mössbauer spectra, is then used to simulate the NRVS spectrum of the related enzyme intermediate.
Influence of calculated NRVS spectra due to truncation of models
Since quantum chemical calculation of a full enzyme is not feasible and we are interested in building a representation of the metal center that is contributing to the NRVS spectrum that can be used to evaluate reactivity through reaction coordinate calculations, it is also necessary to establish a truncation scheme to model the iron active site, while still talking into consideration the effects of the protein. We investigated a series of calculations on a known model compound and the enzyme methane monooxygenase (MMO) alongside several DFT calculations of the full and truncated versions of these examples (see Figure 2)[7][7] Park, K. and Solomon, E. I.: Can. J. Chem. 2014, 92, 975-978.. Model systems which have been truncated in the 1st or 2nd ligand sphere show distinctive intensity and frequency changes compared to the experimental spectra. A reasonable agreement was reached for truncation at the Cα atoms, with replacing the two cut bonds with H atoms and neutralizing the remaining charge by converting the Cα into a methyl group. To avoid artifacts from the methyl vibration and mimic the effects of the protein backbone, the mass of the protons is increased by a factor of 100. This procedure ensures reliable DFT calculations of reasonably-sized clusters, which reproduce the experimentally observed NRVS features and enable reaction coordinate evaluation.
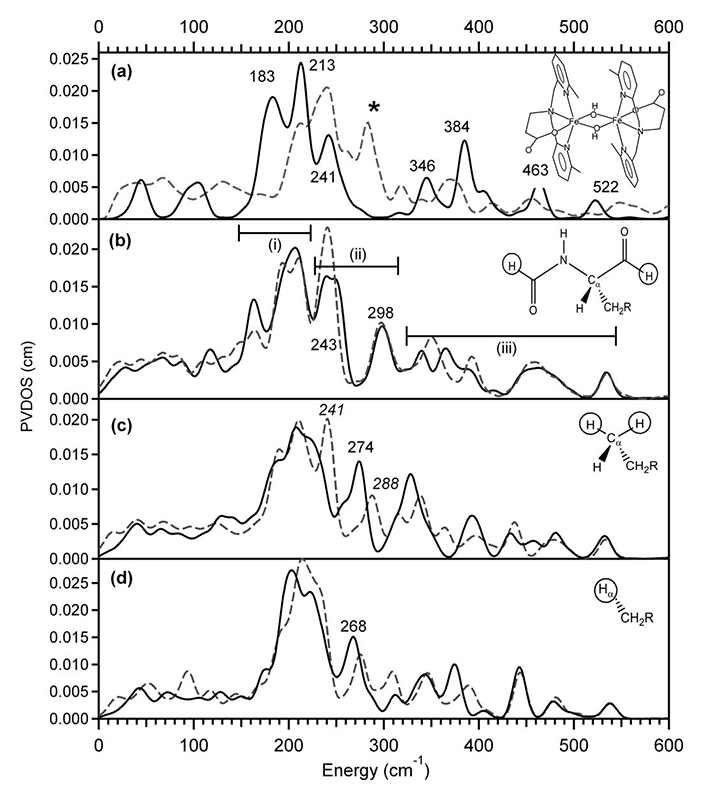
Figure 2 NRVS spectra of (a) [Fe2(μ-OH)2(6Me2-BPP)2]2+ (structure depicted) obtained from experiment (dashed gray line) and DFT computation (solid black line), and the MMO models (b), (c), and (d). The solid black and dashed gray line spectra in (b)-(d) are simulated using constraints with a mass of 1 and 100, respectively.
Results
FeIV=O intermediate in SyrB2
Syringomycin halogenase (SyrB2) is a mononuclear non-heme iron enzyme which, upon reacting with dioxygen, generates a high spin Fe(IV)=O intermediate capable of substrate halogenation (Figure 3). This reactivity is interesting considering that hydroxylation is the thermodynamically preferred chemistry. A number of theoretical studies have suggested mechanisms describing the halogenation of SyrB2; however, these studies are not consistent with experimental data and substrate selectivity. To elucidate the factors governing halo- vs. hydroxylation by SyrB2, our lab has utilized NRVS in conjunction with variable temperature magnetic circular dichroism (VT MCD) to develop an experimentally-calibrated DFT study on reactivity of the iron oxo intermediate with substrates. The geometry of the intermediate, defined with NRVS, possesses a five-coordinate (5C) trigonal bipyramidal structure with the Fe(IV)=O vector axially positioned (Figure 4) and perpendicular to the C-H bond of the substrate[8][8] Wong, S. D.; Srnec, M.; Matthews, M. L.; Liu, L. V.; Kwak, Y.; Park, K.; Bell, C. B., III; Alp, E. E.; Zhao, J.; Yoda, Y.; Kitao, S.; Seto, M.; Krebs, C.; Bollinger, J. M., Jr. and Solomon, E. I.: Nature 2013, 499, 320-323.. The frontier molecular orbitals (FMOs) of this intermediate, which are important for understanding reactivity, have been investigated with VT MCD, revealing a π-FMO that is anisotropic due to the halide equatorialligation that activates this intermediate perpendicular to the Fe-O bond for H-atom abstraction of the substrate C-H bond[9][9] Srnec, M.; Wong, S. D.; Matthews, M. L.; Krebs, C.; Bollinger, J. M., Jr. and Solomon, E. I.: J. Am. Chem. Soc. 2016, 138, 5110-5122.. This produces an HO-FeIII-Cl species with the hydroxyl oriented away from the C● that is perpendicular to the HO-Fe-Cl plane. Marcus theory analysis of the HO-Fe(III)-Cl/C● complex revealed a dπ*Fe-Cl FMO intrinsically more reactive compared to the dπ*Fe-O FMO by ~4 kcal/mol (Figure 5)[10][10] Srnec, M. and Solomon, E. I.: J. Am. Chem. Soc. 2017, 139, 2396-2407.. This energetically favored dπ*Fe-Cl FMO is responsible for lowering the activation barrier of halogenation to be in competition with the thermodynamically favored hydroxylation, resulting in selective chlorination of the native substrate. Halo- or hydroxylation of other substrates depends on the positioning of the radical above the HO-FeIII-Cl plane as shown in Figure 5.
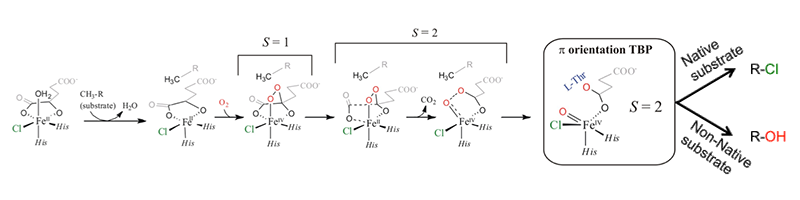
Figure 3 Mechanism of O2 activation by SyrB2[8][8] Wong, S. D.; Srnec, M.; Matthews, M. L.; Liu, L. V.; Kwak, Y.; Park, K.; Bell, C. B., III; Alp, E. E.; Zhao, J.; Yoda, Y.; Kitao, S.; Seto, M.; Krebs, C.; Bollinger, J. M., Jr. and Solomon, E. I.: Nature 2013, 499, 320-323..
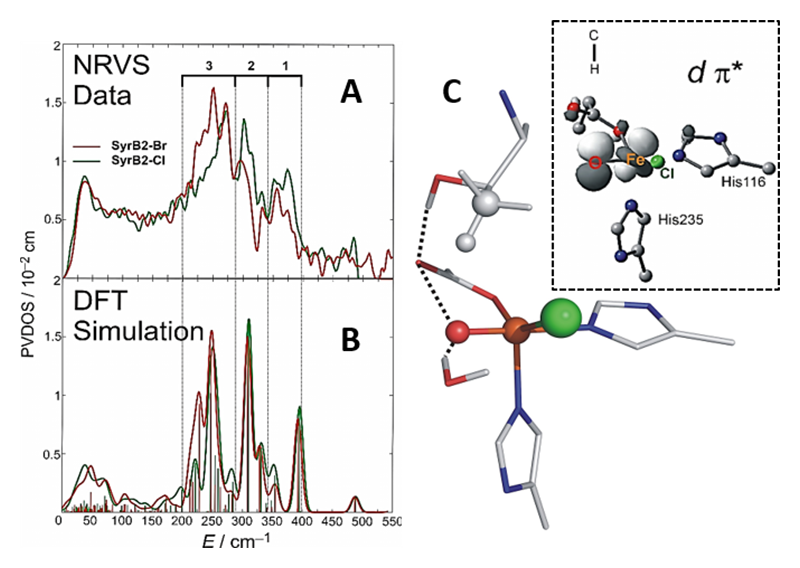
Figure 4 NRVS-derived geometry of the SyrB2 Fe(IV)=O intermediate[8][8] Wong, S. D.; Srnec, M.; Matthews, M. L.; Liu, L. V.; Kwak, Y.; Park, K.; Bell, C. B., III; Alp, E. E.; Zhao, J.; Yoda, Y.; Kitao, S.; Seto, M.; Krebs, C.; Bollinger, J. M., Jr. and Solomon, E. I.: Nature 2013, 499, 320-323.. A: NRVS spectrum of SyrB2 consists of three unique regions of intensity. B: Simulating NRVS data in A with DFT provides. C: The geometry of the Fe(IV)=O intermediate, which is found to be 5C trigonal bipyramidal. Inset: dπ* FMO of the intermediate.
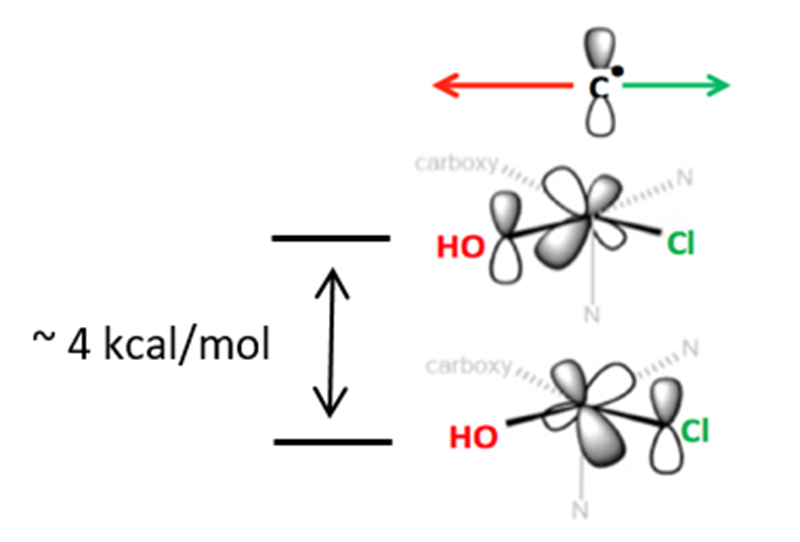
Figure 5 Depiction of dπ*Fe-O and dπ*Fe-Cl FMOs of the SyrB2 HO-Fe(III)-Cl intermediate post H-atom abstraction of the substrate. The ~4 kcal/mol stabilization of the dπ*Fe-Cl FMO permits halogenation of the substrate over the thermodynamically preferred hydroxylation.
FeIII-OOH intermediates
In contrast to the FeIV=O intermediates discussed above, some classes of mononuclear NHFe enzymes utilize FeIII-OO(H) intermediates in their reactivity. These include the anticancer glycopeptide drug bleomycin, which uses a low-spin FeIII-OOH intermediate called activated bleomycin (ABLM) to catalyze the double-strand cleavage of DNA. We used NRVS to determine the structure of this intermediate[11][11] Liu, L. V.; Bell, C. B., III; Wong, S. D.; Wilson, S. A.; Kwak, Y.; Chow, M. S.; Zhao, J.; Hodgson, K. O.; Hedman, B. and Solomon, E. I.: Proc. Natl. Acad. Sci. United States Am. 2010, 107, 22419-22424.. The NRVS spectrum of ABLM (Figure 6A) has a double-peak feature at 398 and 438 cm-1. DFT simulations allowed us to assign this as the pair of degenerate transaxial bends (Figure 6B), split by interaction of the in-plane component with the Fe-OO bend. This splitting was only reproduced computationally for a FeIII-OOH structure and not for the FeIII-O2H2 structure that had been proposed based on calculations, allowing us to experimentally define the geometric structure of this intermediate. From this structure, we calculated the H-atom abstraction reactivity of ABLM, finding that the mechanism proceeded through direct H-atom abstraction by the hydroperoxy rather than through an initial heterolytic cleavage of the O-O bond to form an FeIV=O and ligand radical as in heme chemistry. Our NRVS study thus demonstrated that non-heme FeIII-OOH chemistry is fundamentally different from heme chemistry.
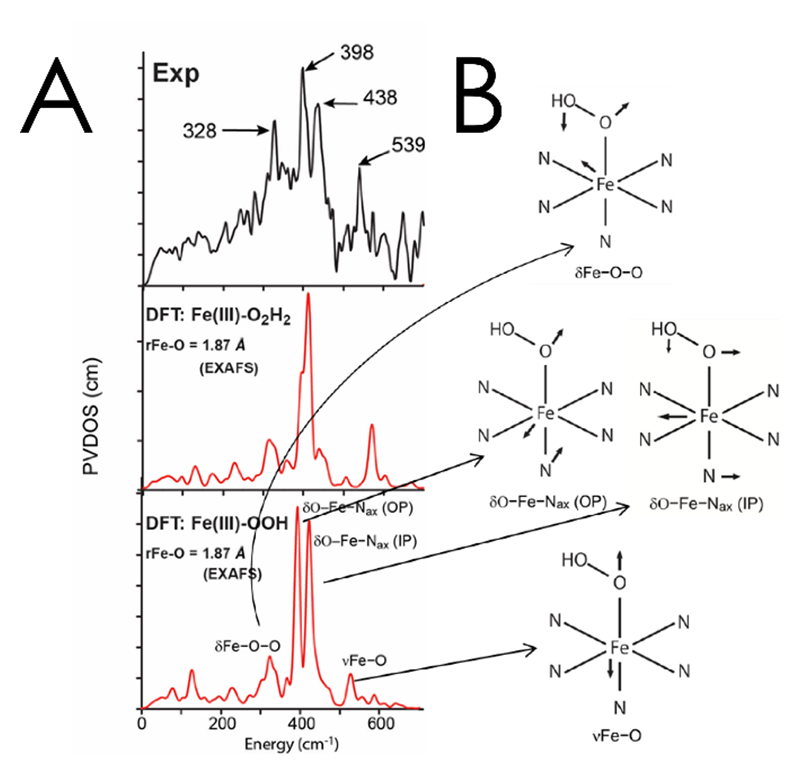
Figure 6 A: NRVS spectrum of ABLM (top) with DFT simulations corresponding to Fe(III)-O2H2 (middle) and Fe(III)-O2H (bottom) structures. B: Schematic depictions of the vibrations assigned to ABLM.
Another class of NHFe enzymes that are proposed to use a FeIII-OOH intermediate in their reactivity are the Rieske dioxygenases (RDOs), which are important in bioremediation. In the RDO benzoate 1,2-dioxygenase (BZDO), an FeIII-OO(H) intermediate (BZDOp) has been trapped, but in contrast to ABLM this is a high-spin ferric-peroxo species. The binding mode and protonation state of this intermediate are unknown, and defining its structure is essential to understanding the cis-dihydroxylation reactivity of these enzymes. We thus expanded our NRVS study of the low-spin FeIII-OOH ABLM to two structurally well-characterized high-spin FeIII-(hydro)peroxy model complexes to obtain NRVS spectroscopic handles for distinguishing between different high-spin FeIII-OO(H) structures: a side-on FeIII-O22- and an end-on FeIII-OOH complex[12][12] Sutherlin, K. D.; Liu, L. V.; Lee, Y.; Kwak, Y.; Yoda, Y.; Saito, M.; Kurokuzu, M.; Kobayashi, Y.; Seto, M.; Que, L., Jr.; Nam, W. and Solomon, E. I.: JACS 2016, 138, 14294-14302.. These data are shown in the top half of Figure 7. The NRVS spectra were assigned using DFT simulations that are consistent with the known structural parameters of these complexes. Using DFT, we systematically correlated between these structures, which defined four spectral handles, shown at the bottom of Figure 7, to differentiate between side-on FeIII-O22-, side-on FeIII-OOH-, and end-on FeIII-OOH- structures. We are now in the process of applying this methodology to analyze NRVS data on BZDOp and to use the geometric and electronic structure determined to evaluate the reaction coordinate of this class of enzymes.
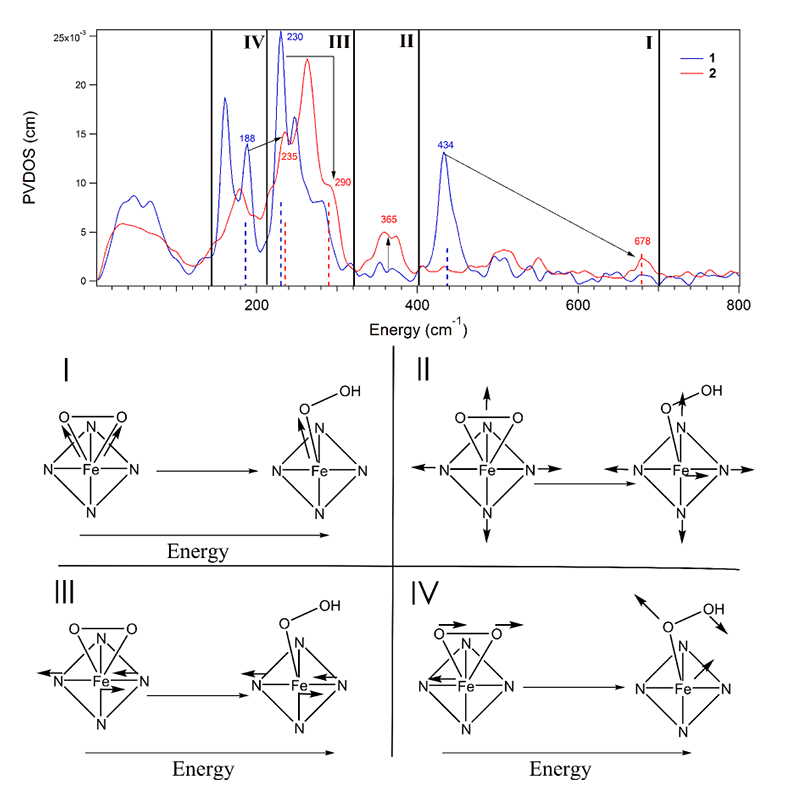
Figure 7 Top: NRVS data for the side-on FeIII-O22- (blue) and end-on FeIII-OOH- (red). The four regions correspond to the four types of vibrations at bottom. Bottom: schematic depiction of the four spectral handles distinguishing between a side-on peroxo and end-on hydroperoxy.
Intermediate P' in AurF
The P' intermediate of 4-aminobenzoate N-oxygenase (AurF) is relatively stable, but directly reacts with 4-aminobenzoic acid to produce 4-nitrobenzoic acid. Thus P' in AurF is a suitable target for defining the nature of a reactive binuclear ferric peroxo intermediate by NRVS.
To determine the structure of P' in AurF, NRVS spectra (Figure 8A) were obtained from samples prepared with 16O2 and 18O2[13][13] Park, K.; Li, N.; Kwak, Y.; Srnec, M.; Bell, C. B.; Liu, L. V.; Wong, S. D.; Yoda, Y.; Kitao, S.; Seto, M.; Hu, M.; Zhao, J.; Krebs, C.; Bollinger, J. M., Jr. and Solomon, E.I.: "Elucidation of the structure and reactivity of the peroxy-level biferric intermediate in AurF." J. Am. Chem. Soc. 2017 doi: 10.1021/jacs.7b02997.. Spectra of these species are divided into three distinct regions: region (1) at energies below 180 cm-1, which is almost featureless, region (2) between 180 cm-1 and 375 cm-1, which displays a series of four peaks with decreasing intensity toward higher energies and region (3) showing two oxygen-isotope-sensitive peaks above 375 cm-1.
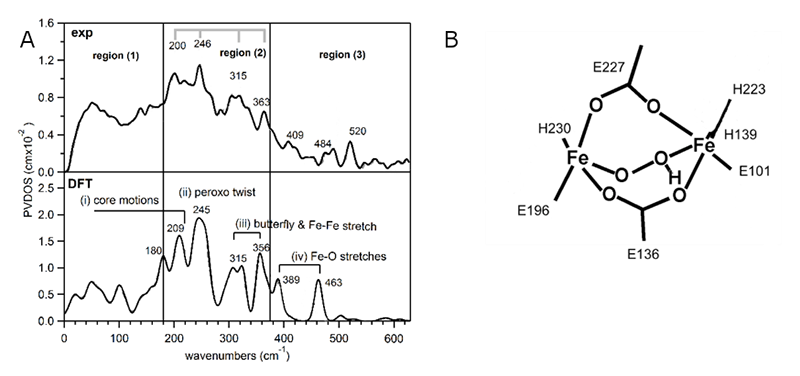
Figure 8 NRVS data and DFT-assisted assignment of P' in AurF. A: (Top) The NRVS spectra of P' obtained with 16O2. (Bottom) The DFT-predicted NRVS spectrum of µ-1,2-hydroperoxo-bridged h3, which is the only structure that reproduces the NRVS data. B: The structure of h3 used to simulate the NRVS data.
In our previous study on µ-1,2-peroxo-bridged Fe(III)2 models, we identified several vibrations associated with the Fe2 core in these regions[14][14] Park, K.; Tsugawa, T.; Furutachi, H.; Kwak, Y.; Liu, L. V.; Wong, S. D.; Yoda, Y.; Kobayashi, Y.; Saito, M.; Kurokuzu, M.; Seto, M.; Suzuki, M. and Solomon, E. I.: Angewandte Chemie Int-Ed 2013, 52, 1294-1298.. To evaluate the NRVS spectrum of P' based on that study, 28 peroxo-bridged structures in different binding modes and with an additional H+ or H2O were calculated. A more detailed assignment of the NRVS spectrum will be presented elsewhere[13][13] Park, K.; Li, N.; Kwak, Y.; Srnec, M.; Bell, C. B.; Liu, L. V.; Wong, S. D.; Yoda, Y.; Kitao, S.; Seto, M.; Hu, M.; Zhao, J.; Krebs, C.; Bollinger, J. M., Jr. and Solomon, E.I.: "Elucidation of the structure and reactivity of the peroxy-level biferric intermediate in AurF." J. Am. Chem. Soc. 2017 doi: 10.1021/jacs.7b02997.; here, we note that only one model, with a µ-1,2-hydroperoxyl bridge, reproduced the experimental spectrum (see Figure 8B).
To examine the viability of this model, the potential energy surface (PES) for the reaction was constructed; the reaction initiates as the O-O bond cleaves and this step triggers oxidation of substrate (Figure 9). This results in a transition state barrier consistent with the reaction rate and a barrier that is ~8 kcal/mol lower in energy than that for the non-protonated peroxo P.
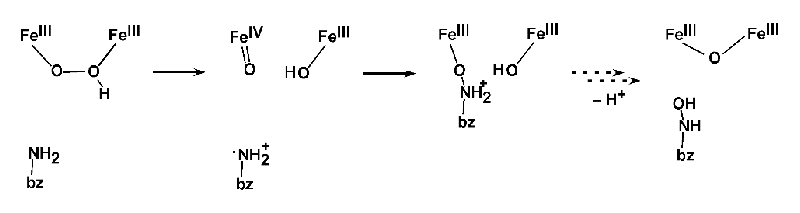
Figure 9 DFT calculations of reaction coordinate for N-oxygenation in AurF.
Protonation increases the electron affinity and stabilizes the high-valent product of O-O bond cleavage. This protonation-enhanced electrophilicity of P' and its conversion to an Fe(IV)Fe(III) intermediate in AurF may be relevant to the activation of P to form P' for the conversion to intermediate X in RNR, and the protonation activation of a peroxo bridged intermediate may also be relevant to the electrophilic chemistry of ToMO. These studies are underway.
Outlook
NRVS has proven to be an invaluable tool for defining the structures of intermediates in non-heme Fe enzymes to elucidate their reactivity. We are now well situated to expand our NRVS studies to other intermediates, including but not limited to BZDOp and FeIV=O heme Compound I and II for mononuclear Fe enzymes and high-valent Q and X intermediates in the NH2Fe enzymes. This will both provide insight into the specific reactivates of these important enzymes, and also into the broader questions regarding the factors governing NHFe versus NH2Fe chemistry, and how heme and non-heme reactivity differ.
Acknowledgments
Beam time at SPring-8 was provided by JASRI under Long Term Proposal 2013B0105-2016A0105. We thank Y. Yoda and M. Seto and his group for their support and assistance in data collection. Financial support for this research was provided to EIS by the National Institutes of Health (GM040392).
References
[1] Solomon, E. I.; Brunold, T. C.; Davis, M. I.; Kemsley, J. N.; Lee, S. K.; Lehnert, N.; Neese, F.; Skulan, A. J.; Yang, Y. S. and Zhou, J.: Chem. Rev. 2000, 100, 235-350.
[2] Solomon, E. I.; Goudarzi, S. and Sutherlin, K. D.: Biochem. 2016, 55, 6363-6374.
[3] Solomon, E. I. and Park, K.: JBIC 2016, 21, 575-588.
[4] Seto, M.; Yoda, Y.; Kikuta, S.; Zhang, X. W. and Ando, M.: Phys. Rev. Lett. 1995, 74, 3828-3831.
[5] Sturhahn, W.; Toellner, T. S.; Alp, E. E.; Zhang, X.; Ando, M.; Yoda, Y.; Kikuta, S.; Seto, M.; Kimball, C. W. and Dabrowski, B.: Phys. Rev. Lett. 1995, 74, 3832-3835.
[6] Sturhahn, W.: Hyperfine Interactions 2000, 125, 149-172.
[7] Park, K. and Solomon, E. I.: Can. J. Chem. 2014, 92, 975-978.
[8] Wong, S. D.; Srnec, M.; Matthews, M. L.; Liu, L. V.; Kwak, Y.; Park, K.; Bell, C. B., III; Alp, E. E.; Zhao, J.; Yoda, Y.; Kitao, S.; Seto, M.; Krebs, C.; Bollinger, J. M., Jr. and Solomon, E. I.: Nature 2013, 499, 320-323.
[9] Srnec, M.; Wong, S. D.; Matthews, M. L.; Krebs, C.; Bollinger, J. M., Jr. and Solomon, E. I.: J. Am. Chem. Soc. 2016, 138, 5110-5122.
[10] Srnec, M. and Solomon, E. I.: J. Am. Chem. Soc. 2017, 139, 2396-2407.
[11] Liu, L. V.; Bell, C. B., III; Wong, S. D.; Wilson, S. A.; Kwak, Y.; Chow, M. S.; Zhao, J.; Hodgson, K. O.; Hedman, B. and Solomon, E. I.: Proc. Natl. Acad. Sci. United States Am. 2010, 107, 22419-22424.
[12] Sutherlin, K. D.; Liu, L. V.; Lee, Y.; Kwak, Y.; Yoda, Y.; Saito, M.; Kurokuzu, M.; Kobayashi, Y.; Seto, M.; Que, L., Jr.; Nam, W. and Solomon, E. I.: JACS 2016, 138, 14294-14302.
[13] Park, K.; Li, N.; Kwak, Y.; Srnec, M.; Bell, C. B.; Liu, L. V.; Wong, S. D.; Yoda, Y.; Kitao, S.; Seto, M.; Hu, M.; Zhao, J.; Krebs, C.; Bollinger, J. M., Jr. and Solomon, E.I.: "Elucidation of the structure and reactivity of the peroxy-level biferric intermediate in AurF." J. Am. Chem. Soc. 2017 doi: 10.1021/jacs.7b02997.
[14] Park, K.; Tsugawa, T.; Furutachi, H.; Kwak, Y.; Liu, L. V.; Wong, S. D.; Yoda, Y.; Kobayashi, Y.; Saito, M.; Kurokuzu, M.; Seto, M.; Suzuki, M. and Solomon, E. I.: Angewandte Chemie Int-Ed 2013, 52, 1294-1298.




