



Volume 29, No.2 Pages 94 - 98
1. 最近の研究から/FROM LATEST RESEARCH
タンパク質構造とAIによる新規胃酸抑制剤候補化合物のデザイン
AI-driven De novo Drug Design Based on Protein Structures
北海道大学 大学院理学研究院化学部門 分子生命化学研究室 Department of Chemistry, Faculty of Science, Hokkaido University
- Abstract
- 胃プロトンポンプを標的とした胃酸抑制剤は、胃潰瘍や逆流性食道炎の治療、胃がんの主原因とされるピロリ菌の除菌時の胃酸抑制を目的として用いられる。その世界市場規模は2兆円とも言われ、巨大なマーケットを持つ薬剤である。我が国でも、これまでに複数の薬剤が上市され治療に用いられているが、これらはすべてフェノタイプスクリーニングによって開発されたものであり、その結合構造は未知であった。我々は2018年に胃酸抑制剤の結合構造解析に初めて成功して以来、これまで7つの異なる薬剤/阻害剤の結合構造を明らかにしてきた。本稿では、これら結合構造の情報を、AIを利用することで阻害剤のデザインへと応用した例を紹介する。
1. はじめに
不快な胸やけに襲われたとき、ふと思い起こすのは、我々の胃袋がpH1もの強い酸性環境にさらされているという事実である。この過酷な環境は、胃プロトンポンプ(H+、K+-ATPase)と呼ばれる膜タンパク質が、胃の内部にH+を能動輸送することによって作り出される。通常は胃粘膜が胃壁を保護しているが、暴飲暴食、内的外的ストレスによって胃酸と胃粘膜分泌のバランスが崩れると、胃酸が自身を傷つけてしまうことで、重篤な場合には胃潰瘍や逆流性食道炎といった症状を呈する。また、胃がんの主原因とされるピロリ菌の除菌時には、抗生物質の薬効を担保するために胃内部の酸性度を軽減する必要がある。このためには、単純明快、胃酸の分泌を止めてやるのが効果的である。したがって、胃酸分泌の直接の担い手である胃プロトンポンプが胃酸抑制剤のターゲットとされたのは、至極当然といえよう[1][1] J. M. Shin and G. Sachs: Curr. Gastroenterol. Rep. 10 (2008) 528.。オメプラゾールに代表されるProton Pump Inhibitor(PPI)は、胃酸に関連した病態の治療に広く用いられ、大きな成功を収めた。PPIは、服用時点では不活性な分子であるが、胃に到達し酸性環境によってリアクティブな化合物へと変換されるという特徴を持つ。その結果、プロトンポンプに保存されたシステイン残基(Cys813)と共有結合を形成し、これを不活性化する[2][2] J. M. Shin, Y. M. Cho and G. Saches: J. Am. Chem. Soc. 126 (2004) 7800.。しかしながら、酸性で活性化されるということは、逆に言えば化合物としては酸性状態で不安定ということである。そのためPPIは腸溶剤として処方され、最大効用まで1日程度の時間を要する。つまり、「気持ち悪い!」と思ってPPIを服用しても、すぐには効かないわけである。また、PPIは遺伝子多型のある肝臓の薬物代謝酵素(CYP2C19)によって分解されるので、効き目に個人差があり、より迅速かつ確実な治療を目指して新しいクラスの薬剤が開発されてきた。2015年に我が国で上市されたvonoprazan(商品名:タケキャブ、武田薬品工業)は、その先駆けであり、胃プロトンポンプを直接ブロックすることで、迅速な治癒を実現した[3][3] K. Otake et al.: Adv. Ther. 33 (2016) 1140.。Vonoprazanの他にも、インドや中国で上市されているrevaprazan[4][4] K. S. Han et al.: Biopharm. Drug. Dispos. 19 (1998) 493.や、韓国で成功を収め我が国でも臨床試験が進んでいるtegoprazan[5][5] D. K. Kim et al.: J. Pharmacol. Exp. Ther. 369 (2019) 318.、他にも毒性のために薬剤としては利用されていないが、最初に特異的阻害剤として合成されたプロトタイプ化合物ともいえるSCH28080[6][6] J. J. Kaminski et al.: J. Med. Chem. 28 (1985) 876.をはじめ、多くの阻害剤が存在する[1][1] J. M. Shin and G. Sachs: Curr. Gastroenterol. Rep. 10 (2008) 528.。これら新しいクラスの薬剤は、H+、K+-ATPase活性をH+の対向輸送イオンであるK+競合的に阻害することから、Potassium-Competitive Acid Blockers(P-CAB)と呼ばれる(図1)。
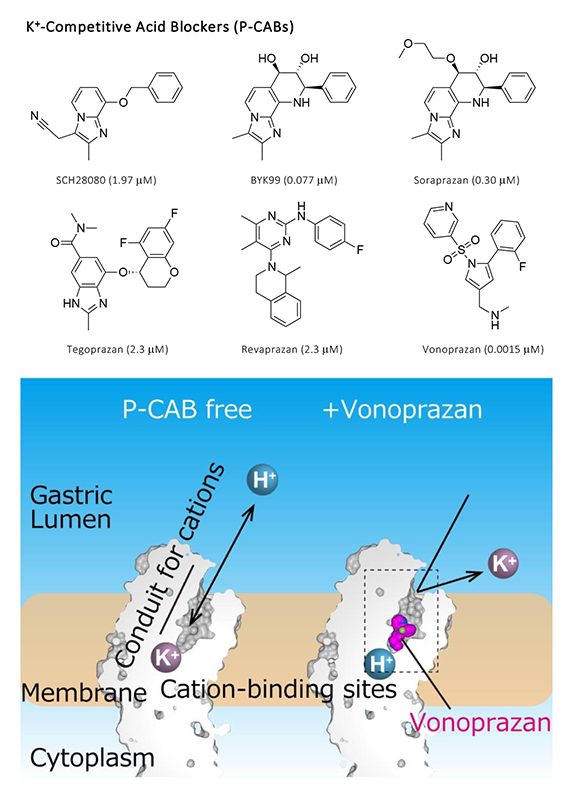
図1 P-CABの化学構造とその作用機序
(上)これまで構造解析されたP-CABの化学構造(括弧内はIC50)。
(下)胃プロトンポンプの膜貫通領域の断面図。P-CAB(例としてvonoprazanを示す)は、カチオン結合サイトと胃内腔を繋ぐ通路に結合することで、文字通りカチオン輸送をブロックする。
このように、現在では胃酸抑制剤として複数の薬剤が利用できる状態にあるが、すべての薬剤はフェノタイプスクリーニングによって開発されたものであり、それらの結合構造は明らかではなかった。これがわかれば、構造に基づいた論理的なデザインによって現行の薬剤の改良や、新規薬剤の開発に結び付くはずである。我々はこれまでに放射光を利用したX線結晶構造解析、およびクライオ電子顕微鏡(CryoTEM)による単粒子解析によって、複数の薬剤や阻害剤の結合構造を決定してきた[7-10][7] K. Abe, K. Tani and Y. Fujiyoshi: Nat. Commun. 2 (2011) 155.
[8] K. Abe, K. Irie, H. Nakanishi, H. Suzuki and Y. Fujiyoshi: Nature 556 (2018) 214.
[9] K. Abe, K. Yamamoto, K. Irie, T. Nishizawa, A. Oshima: Nat. Commun. 12 (2021) 5709.
[10] S. Tanaka et al.: J. Med. Chem. 65 (2022) 7843.。
2. 薬剤結合構造
これまで構造解析されたすべてのP-CABは、胃内腔に面した、カチオン結合サイトと繋がるポケット内に結合していたが、化合物の結合様式にはいくつかの共通点と相違点が見られた(図2)。SCH28080に代表される化学骨格を持つ化合物群は、ポケットの胃管腔側に偏って結合していた。一方で、vonoprazanは、第二級アミンをカチオン結合サイト近傍に配置する形でポケットの奥深く結合していた。Revaprazanは、ちょうどSCH28080とvonoprazanの中間の位置に結合していた[9][9] K. Abe, K. Yamamoto, K. Irie, T. Nishizawa, A. Oshima: Nat. Commun. 12 (2021) 5709.。個々の化合物/薬剤の結合状態は、アミノ酸レベルで詳細に理解されたが、次の課題はこれらの構造情報をドラッグデザインに活用することである。単純なアイデアとしては、化合物の結合場所が複数存在するならば、それらすべてに結合できる化合物を開発することで、より親和性の高いものができるのではないか、というものである。
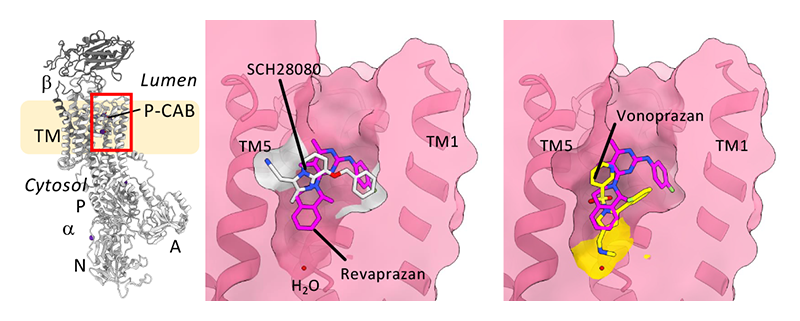
図2 P-CABの区別できる結合モード
胃プロトンポンプの構造(左)。赤枠で囲った部分が阻害剤の結合ポケット(右側にその拡大図を示す)。SCH28080(白)は白く示した位置を中心に結合しているが、vonoprazan(黄色)はポケットの奥深くまで到達している。Revaprazan(マゼンタ)は、これらの中間的な結合様式を示す。
3. AIによるドラッグデザイン
構造を見てすぐに思いつくのは、元となる化合物に何らかの官能基を修飾することで、複数の結合サイトを橋渡しすることであろう。しかしながらこの方法では、基本的な化学骨格が同じであるために、オリジナル化合物の特許が薬剤としての開発の障害となる。また、類似の化学骨格を用いる場合、薬物動態の大幅な変化を期待するのは難しいであろう。つまり既存の薬剤結合構造を参考にしつつ「De Novo化合物」をデザインする必要があるわけである。これは人間にとって(少なくとも経験豊富な創薬化学者などではない著者には)、そう簡単なものではない。一体どういう化学構造で複数の結合サイトを繋げばいいかというのが、次の問題である。
我々はこの問題にAIを利用した。理論創薬研究所の吉森博士が開発したAIプラットフォーム「Deep Quartet」(DQ)[11][11] A. Yoshimori, E. Kawasaki, C. Kanai and T. Tasaka: Chem. Pharm. Bull. 68 (2020) 227.は、bio-activeな化合物の構造(ChEMBL、https://www.ebi.ac.uk/chembl/)を事前学習し、タンパク質の構造の上に任意に指定したファーマコフォアフィーチャーを満たすde novo化合物をデザインすることができる(図3)[12][12] K. Abe et al.: Commun. Biol. 6 (2023) 956.。
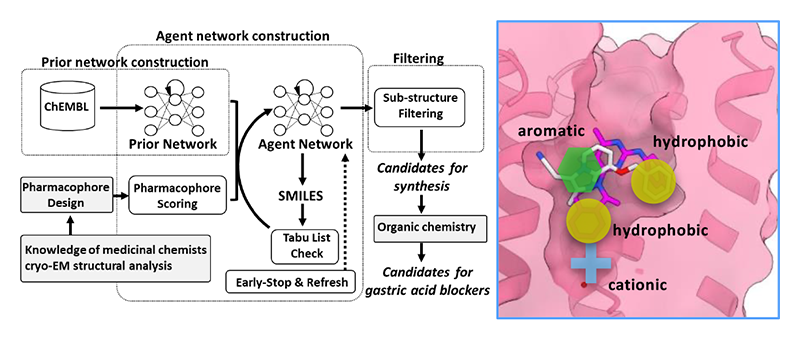
図3 Deep Quartetによるde novo drug design
Deep Quartet(左)は、データベース(ChEMBL)に格納されたbio-activeな化学構造を事前学習し、タンパク質の構造上に座標として指定したファーマコフォアフィーチャー(右)を満たすde novo化合物を生成する。詳しくは文献[12][12] K. Abe et al.: Commun. Biol. 6 (2023) 956.を参照のこと。
4. First trial – ほんとにこれでいいの?
まず初めに、SCH28080の結合構造をテンプレートとした計算を行った[10][10] S. Tanaka et al.: J. Med. Chem. 65 (2022) 7843.。P-CAB結合サイトに4つのファーマコフォアフィーチャーを指定し(図3)、DQによってこれを満たす複数のde novo化合物の候補を得た(図4)。初めてこれらの化合物の構造を見たときの正直な感想は「?」である。これらの候補の大部分は、細長いアルキン(炭素―炭素間の三重結合)で連結された、薬剤としてあまり目にすることがない化学構造であった。ともかく複数の候補の中から、有機合成の難易度や人の目で選別したものを実際に合成し、H+、K+-ATPase活性を指標とした阻害能を検討したところ、2番目に合成した化合物(DQ-02)が、弱いながらも阻害活性を示した。Cryo-EM(EM01CT@SPring-8)によって解析したDQ-02結合構造は、多くの水を解像できる分解能(2.1 Å)であるにも関わらず、DQ-02に相当する密度は非常に乱れていた。これはおそらく、アルキン部分の細い構造がポケット内で安定に結合するには細すぎるために、結合様式が一様ではないことが原因であると考えられ(図ではDQ-02の2つの結合モードをモデルしている)、明らかに高親和性の結合には不向きであると考えられた。
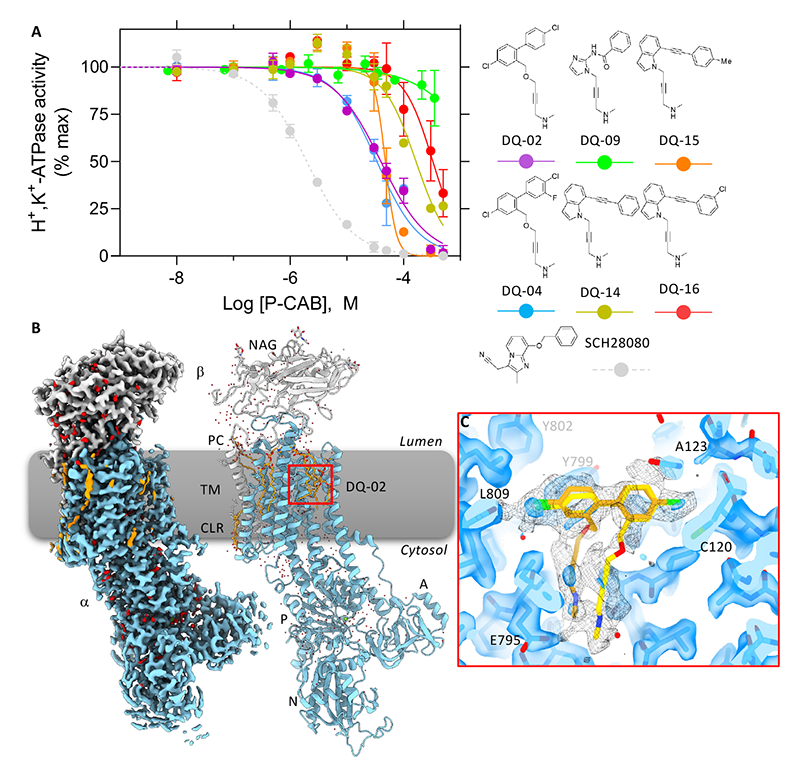
図4 DQ-02結合構造
(A)H+、K+-ATPase活性阻害の濃度依存性。
(B)DQ-02が結合した胃プロトンポンプのcryoTEM構造。
(C)DQ-02結合部位の拡大図。タンパク質の密度を水色で、DQ-02に相当する密度をmeshで示す。図中では2種類のDQ-02結合モードをモデルした。
では、なぜそのような化学構造がデザインされてしまったのであろうか?原因は、言ってみればヒューマンエラー(著者のせい)であった。最初の計算に用いたSCH28080結合構造(5ylv)[7][7] K. Abe, K. Tani and Y. Fujiyoshi: Nat. Commun. 2 (2011) 155.は、他の薬剤結合構造(例えばvonoprazan結合構造、5ylu)よりも、結合ポケットの幅が狭いことがわかる(図5)。この狭いポケットに結合できる化学構造を、DQは言われた通りに計算しただけであって、この構造テンプレートを選んだこと自体に問題があったわけである。そこで今度は、ポケットの幅が広いvonoprazan結合構造[7][7] K. Abe, K. Tani and Y. Fujiyoshi: Nat. Commun. 2 (2011) 155.をテンプレートして、もう一度同様の計算を行うことにした。
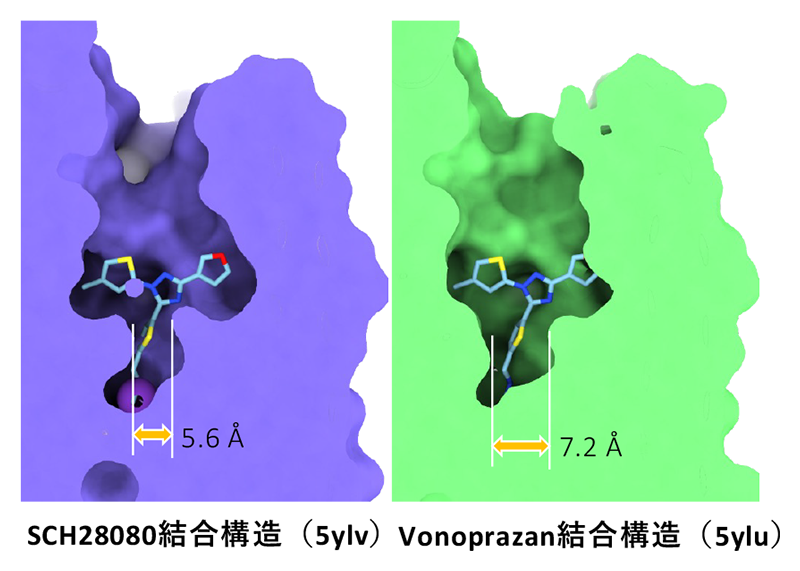
図5 構造テンプレートの違い
SCH28080結合構造(左)では、vonoprazan結合構造(右)と比べて結合ポケットの一部が狭くなっている。
5. Second trial – それっぽい化合物
Vonoprazan結合構造をテンプレートとした再計算の結果、今度は複数の環構造をもつ「drug-like」な化学構造が多く得られた。これらの中からシンプルなものを化学合成し、その阻害能を検討したところ、6番目の化合物(DQ-06)が、基準となるSCH28080よりも高い親和性を示した。CryoTEM(2.19 Å)によってDQ-06は明瞭に解像され、その結合状態を検討できる構造が得られた。
6. 構造に基づいた改良
想定通り、DQ-06は指定した複数のファーマコフォアフィーチャーを満たすものであった(図6)。DQ-06の第二級アミンは、vonoprazanがそうであるように、カチオン結合サイト近傍に位置し、中央の環構造は他のP-CABと同様にTyr799とπ結合を形成、誌面右側に位置するベンゼン環はSCH28080とその類似化合物のようにA123近傍に結合している。しかしながら、より詳細に構造を見ていくと、2番目の環と結合ポケットの間に若干の隙間があることが分かった(図6)。
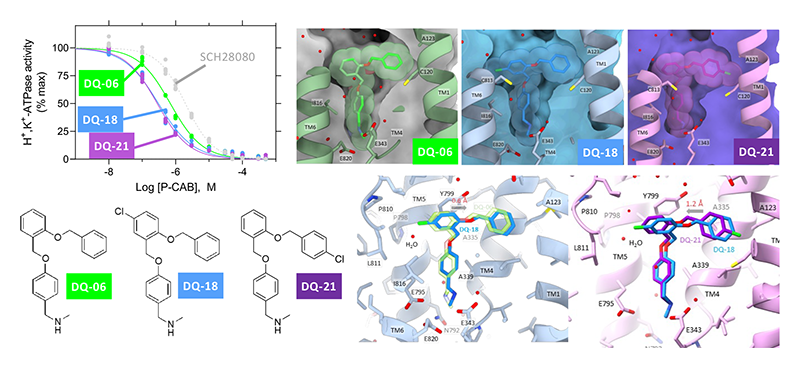
図6 DQ-06とその類縁化合物
DQ-06およびその類縁化合物によるH+、K+-ATPase活性阻害の濃度依存性(左)と、それぞれの結合部位の構造(右上)を示す。結合構造比較(右下)により、DQ-18はDQ-06と比べて、Clの導入によって右側に0.6 Åシフトし、DQ-21はDQ-18と反対側の位置にClを導入したことによって左側に1.2 Åシフトしていることがわかる。
DQ-06を改良し、結合ポケットの形にフィットさせることができれば、親和性の向上が期待できるであろう。そこで、構造上に見出した隙間の位置近傍に塩素(Cl)を導入した化合物(DQ-18)を合成した。活性測定の結果は、予想通り若干の親和性改善が見られた。DQ-18の結合構造(2.10 Å)から、想定した位置にClが配置されることで隙間を埋めていることが判明した。
DQ-18で得られた親和性の向上が、結合ポケットとの隙間と関係しているのであれば、化合物の反対側にClを導入しても同様のことが起こるはずである。この仮説を検証するために、DQ-21を合成した。阻害能はDQ-18とほぼ同じであり、結合構造も想定通りであった。
特筆に値するのは、cryoTEMによる構造解析の精度である。DQ-06にClを導入することで、DQ-18は0.6 Åだけ誌面右側にずれるが、反対側にClを導入したDQ-21は、DQ-18と比較して1.2 Å左側にずれて結合している(つまりDQ-06を基準とすると0.6 Å左側ということになる)。この精度の構造解析が達成されたことは、cryoTEMによる構造がドラッグデザインに十分応用できることを物語っている。
7. おわりに
本稿で紹介した任意のファーマコフォアを満たす化合物をAIによってデザインする枠組みは[12][12] K. Abe et al.: Commun. Biol. 6 (2023) 956.、胃プロトンポンプだけではなく、薬剤結合構造が既知の様々なタンパク質への応用が期待できる。
本研究はBL32XU、BL41XU、BL45XUを利用したX線結晶構造解析、およびEM01CTを利用したcryoTEM単粒子解析(SPring-8利用研究課題No. 2017B2701、2018B2703、2020A2707、2021B2530、2022B2522、2023B2518)によって実施しました。多くの共同研究者の方々、ビームラインサイエンティストの方々にこの場を借りて感謝申し上げます。
参考文献
[1] J. M. Shin and G. Sachs: Curr. Gastroenterol. Rep. 10 (2008) 528.
[2] J. M. Shin, Y. M. Cho and G. Saches: J. Am. Chem. Soc. 126 (2004) 7800.
[3] K. Otake et al.: Adv. Ther. 33 (2016) 1140.
[4] K. S. Han et al.: Biopharm. Drug. Dispos. 19 (1998) 493.
[5] D. K. Kim et al.: J. Pharmacol. Exp. Ther. 369 (2019) 318.
[6] J. J. Kaminski et al.: J. Med. Chem. 28 (1985) 876.
[7] K. Abe, K. Tani and Y. Fujiyoshi: Nat. Commun. 2 (2011) 155.
[8] K. Abe, K. Irie, H. Nakanishi, H. Suzuki and Y. Fujiyoshi: Nature 556 (2018) 214.
[9] K. Abe, K. Yamamoto, K. Irie, T. Nishizawa, A. Oshima: Nat. Commun. 12 (2021) 5709.
[10] S. Tanaka et al.: J. Med. Chem. 65 (2022) 7843.
[11] A. Yoshimori, E. Kawasaki, C. Kanai and T. Tasaka: Chem. Pharm. Bull. 68 (2020) 227.
[12] K. Abe et al.: Commun. Biol. 6 (2023) 956.
(現所属)北海道大学
大学院理学研究院化学部門 分子生命化学研究室
〒060-0810 札幌市北区北10条西8丁目
TEL : 011-706-3505
e-mail : kabe@sci.hokudai.ac.jp




