



Volume 29, No.1 Pages 14 - 18
1. 最近の研究から/FROM LATEST RESEARCH
XFELと電子線による低分子有機化合物の微小結晶構造解析-二線源の特性を生かし、水素原子と電荷に関する情報を取得-
XFEL and electron microcrystallography of small organic compounds -measuring charges and chemical bonds of hydrogen atoms using the two beam sources-
[1](国)理化学研究所 放射光科学研究センター RIKEN, SPring-8 Center、[2]東北大学 多元物質科学研究所 Tohoku University IMRAM
- Abstract
- 複雑な構造を持つ有機化合物では、X線回折に適したサイズの結晶の調製が難しいことがある。そこで、創薬、材料科学などの広範な分野で、小さな結晶から原子配置を決定することが重要になっている。私たちは、この問題の解決のために、X線自由電子レーザーを用いた低分子有機化合物の微小結晶構造解析法を開発した。実証実験では有機蛍光分子であるローダミン6Gの構造を、0.82 Åの空間分解能で決定できた。電子回折から得た構造と比較すると、どちらの構造も水素原子の位置を化学結合の種類に依存して正確に区別できることが分かった。両構造間の水素原子の測定距離の違いは、X線と電子線の散乱の特性を反映しており、また、原子座標の信頼性はXFELの方が高いが、電子線は電荷に対してより高い感度を示すことも明らかにできた。
1. はじめに
有機合成化学、薬学、材料科学などの分野において、研究対象となる低分子有機化合物の原子の立体配置は欠くことのできない情報である。しかし、従来のX線回折に適した数百μm以上の大きさの結晶作製が困難であることも多い。構造決定に必要な結晶サイズを決める原子の散乱断面積は、X線と電子線で顕著に異なり、電子線ではX線に比べて4から5桁も大きい[1][1] Henderson, R.: Q. Rev. Biophys. 28 (1995) 171-193.。この性質を反映し、X線回折にサイズが満たない厚さ数百nm以下の結晶から、電子回折像が得られる。電子顕微鏡を用いて試料を回転させながら回折像を連続的に記録する電子線三次元結晶構造解析法[2, 3][2] Nannenga, B. L., Shi, D., Leslie, A. G. W. and Gonen, T.: Nat. Methods 11 (2014) 927-930.
[3] Yonekura, K., Kato, K., Ogasawara, M., Tomita, M. and Toyoshima, C.: Proc. Natl. Acad. Sci. U. S. A. 112 (2015) 3368-3373.により、原子レベルの空間分解能で結晶構造の決定が可能になった。逆に、電子線の透過力は低く、厚い結晶への電子回折の適用は著しく制限される。
X線自由電子レーザー(XFEL)施設SACLAは、高強度かつ極短いX線パルスを発生させる。1パルスで試料破壊前のデータ収集が可能であり[4][4] Barty, A. et al.: Nat. Photonics 6 (2012) 35-40.、電子線の散乱断面積との大きな差を補うことができると考えられた。XFELパルスを用いた連続結晶X線構造解析(SX)では、多数の微小結晶から回折パターンを記録する。この手法は、これまで主にタンパク質結晶に適用されてきた[5, 6][5] Chapman, H. N., et al.: Nature 470 (2011) 73-78.
[6] Boutet, S. et al.: Science 337 (2012) 362-364.が、それは、低分子有機化合物の結晶の小さな結晶格子では、カメラ1フレーム当たりの回折点の数が少なくデータ処理が難しいという理由からである。
それでも、2022年には、無機-有機ハイブリッド材料の小さな結晶から、1.2~1.35 Åの分解能の構造がXFELを用いて決定されたことが報告された[7][7] Schriber, E. A. et al.: Nature 601 (2022) 360-365.。同研究では、微小結晶試料を含む溶液を細いノズルの先端から噴出させることで、XFELパルスを照射する手法がとられた。この試料供給法は、タンパク質結晶のSX解析で最も頻繁に用いられているものであるが、低分子でよくみられる試料形態の微結晶粉末などに対して、適しているとは考えられなかった。また、回折点の指数付けには、回折像を円環平均し粉末パターンを模した一次元の強度分布が用いられたが、私たちは別のアプローチをとった。解析された試料にはAg、Se、またはTeなどの重原子が含まれ回折強度の向上に寄与していたにも関わらず、得られた構造の分解能は水素原子の解像に不十分であった。
有機分子構造において、水素原子は分子内・分子間で広範に非共有結合を形成し、分子の性質、機能に大きな影響を及ぼす。しかし、X線や電子線で得られる水素原子からの信号は弱く、1 Åよりも良い空間分解能での高精度な構造解析が不可欠である。これに対し、中性子回折は水素原子の検出に向き、その原子核の位置を決定できるが、現行の中性子源から十分な強度の中性子線が得られないという問題がある。解析にはmmサイズの巨大な結晶が必要になってしまう。
中性子線は核に散乱されるのに対して、X線は原子核の周りの電子、電子線は原子の周りのクーロンポテンシャルによってそれぞれ散乱される。このように、3つの代表的な量子線の散乱実体は大きく異なる。そこで、同じ微小結晶試料からXFELと電子線で得られた構造を詳しく比較し、実験的信頼性を評価する必要があった[8][8] Takaba, K. et al.: Nat. Chem. 15 (2023) 491-497.。
2. 構造解析
X線の散乱が少ないポリイミド製の4 mm × 4 mmの試料支持板をデザインし、その表面にローダミン6Gの微小結晶を流動パラフィンと共に散布し試料とした。微小結晶の平均サイズは2.5 μmであった(文献[8]の補足図1)。この試料支持板を高速で移動させ、径約1 μmに集光した15 keVの高エネルギーXFELパルスを、10 μmの間隔で個々の微小結晶に照射し、回折パターンを集めた(図1)。パルス長は約7 fs、パルス間隔は30 Hzで、測定は大気中、室温で実施した。支持板一つあたり約1.5時間の測定から13万枚以上の回折像が得られ、このうち約6万5千枚が、結晶からの反射を確認できる“ヒット”パターンに分類された。図2(a)に支持板上の微小結晶の分布を示す。
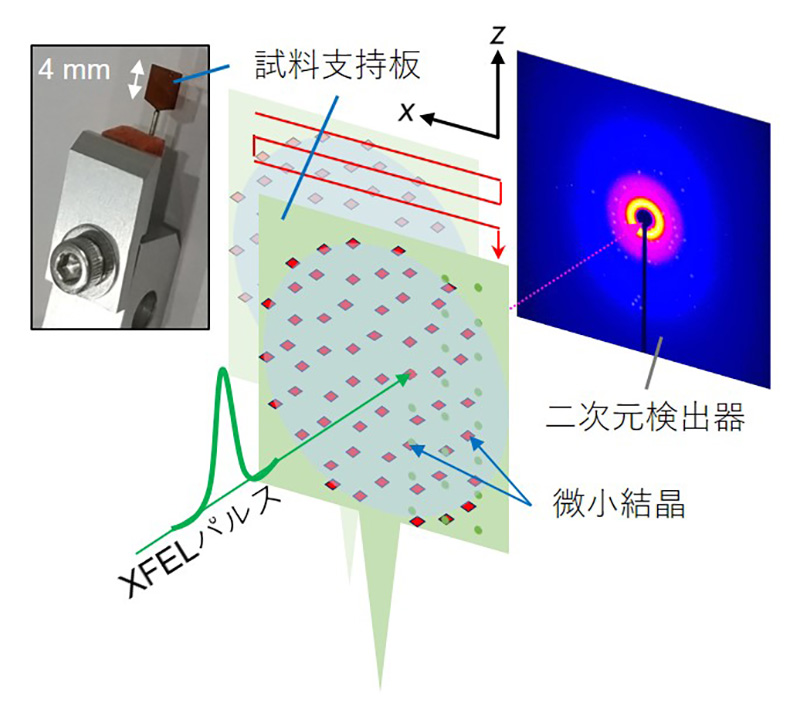
図1 X線自由電子レーザー(XFEL)による回折データ測定の模式図。
4 mm × 4 mmの大きさの試料支持板に微小結晶を散布し、高速に移動させながら(赤矢印)、XFELパルスを1秒間に30回照射し、回折パターンを後方の検出器で記録する。文献[8]より改変。
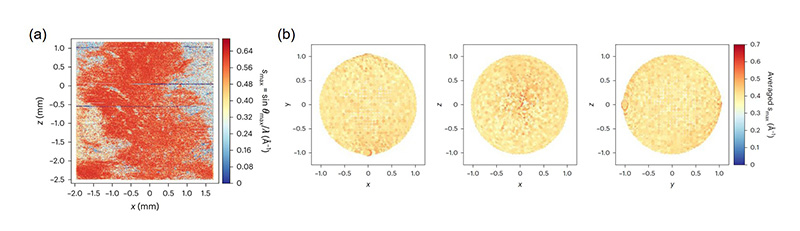
図2 試料支持板上の微小結晶の分布と向き。
(a) ローダミン6Gの微小結晶の分布を、その場所から得られた回折フレーム中の回折点の最高分解能(smax)で色分けし表示。smax = sinθmax/λと定義され、θmaxは回折点の最大散乱角の半分の値を表し、λはX線の波長に対応する。(b) 指数付けされたフレームに寄与する結晶方位を、逆格子の単位ベクトルa*の端点に色付けし表示。各点のサイズと色は、対応する方位をとるフレーム数(結晶数)と平均smaxにそれぞれ対応する。図は左からx–y平面、x–z平面、y–z平面への射影で、入射X線の方向はy軸に、x軸及びz軸は支持板平面上の直行座標を表す(図1)。文献[8]から転載。
XFELと並行し、加速電圧300 kVのクライオ電子顕微鏡(日本電子CRYO ARM 300)を用いて、同じ試料の複数の微小結晶から、試料ステージを回転させ電子回折像を集めた[9, 10][9] Yonekura, K., Ishikawa, T. and Maki-Yonekura, S.: J. Struct. Biol. 206 (2019) 243-253.
[10] Takaba, K., Maki-Yonekura, S., Inoue, S., Hasegawa, T. and Yonekura, K.: Front. Mol. Biosci. 7 (2021) 612226.。得られた回転回折データセットをクラスター解析で分け[10][10] Takaba, K., Maki-Yonekura, S., Inoue, S., Hasegawa, T. and Yonekura, K.: Front. Mol. Biosci. 7 (2021) 612226.、40-50セットを統合し構造決定した。
XFELの回折像は、電子線で得られた結晶格子情報を与えることで指数付けでき、その成功率はタンパク質のSXパターンに匹敵する69.2%に達した。以上の測定と回折像の処理の手順は、前出の無機-有機ハイブリッド材料で採用された液体噴霧を用いる方法に比べ、約20倍高効率であった。抽出された反射強度はモンテカルロ法により積分し、統合した。統合したデータから、支持板上に分散した結晶の向きはランダムであり(図2(b))、すべての反射点が記録されていることが確認された。統合したデータをab initio法で位相付けし、0.82 Åの空間分解能でローダミン6Gの構造を決定することに成功した(図3)。実験データから水素原子を省いたモデルの構造因子を差し引いた差フーリエマップによって水素原子の密度が明瞭に解像された(図3(b), (c)、図4(b))。差マップ内の原子間の残留密度は、共有結合電子に相当すると考えられる(図4(b))。
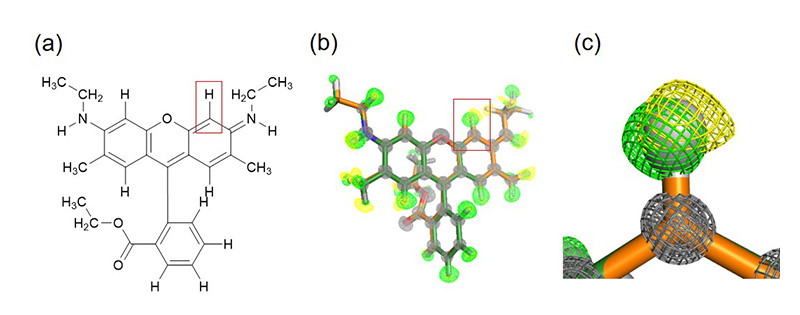
図3 XFEL及び電子線を用いて決定した結晶中のローダミン6Gの立体構造。
(a) ローダミン6Gの構造式。(b) 結晶構造と水素原子分布の重ね合わせ。緑はXFEL、黄色は電子線での測定データからそれぞれ得られた水素原子の密度を示す。水素原子は、XFELでは結合相手の原子に近い位置に観察される。水素以外の原子位置(灰色)は両者の構造間でほぼ等しい。(c) (a)と(b)の赤枠で囲った部分の拡大。
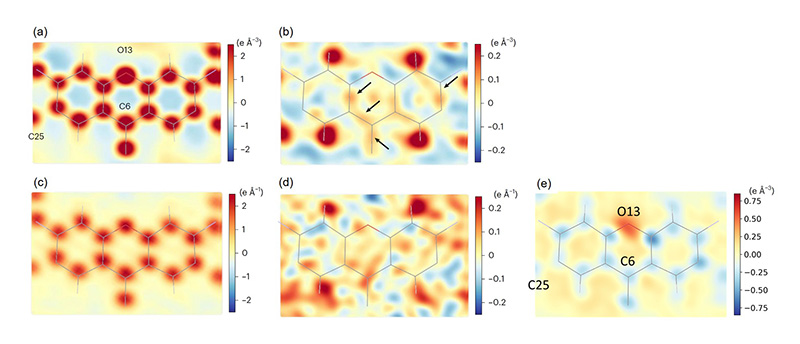
図4 ローダミン6Gの3つの六員環から構成されるキサンテン環平面での電子密度とクーロンポテンシャルマップの二次元断面。
(a) XFELから得られた実験振幅(Fo)マップの断面に原子モデルを重ねて表示。見やすいよう、いくつかの原子にラベル付けした。(b) XFELの実験データと水素を除いた計算データの差マップ(mFo-DFc)の断面。図中の矢印は、共有電子に対応する残留密度を表すと考えられる。(c, d) 電子回折から得られた(a)と(b)に対応する断面。色の表示は、各マップの右側に示されているグラデーションバーに従う。差マップの残留密度(b)と(d)の比較では、後者にノイズが多くみえる。(e) (c)のクーロンポテンシャルを電子密度に変換し計算した差マップ。文献[8]より改変。
3. 精度の比較
電子回折からは0.90 Åの分解能で構造が得られ、差マップから水素原子が可視化できた。XFELと電子回折から得られた両構造間で、水素以外の原子位置はほぼ一致した。一方、実験データとモデルの不一致を示すR値はXFELの回折データが優り、これを反映して結合長の標準不確かさも約1/5~1/2になった[8][8] Takaba, K. et al.: Nat. Chem. 15 (2023) 491-497.。このように電子回折データの品質が劣ることは、標準的なX線結晶構造解析との比較からも、一般的傾向として報告されている[11][11] Clabbers, M. T. B., Gruene, T., van Genderen, E. and Abrahams, J. P.: Acta Crystallogr. A 75 (2019) 82-93.。前述のように、電子線の試料への透過力は低く、特に試料の傾斜角が大きくなる際に試料の厚みは増加し、構造解析の大きな制限になる。また、多重散乱や部分電荷に対する電子散乱の取り扱いなど、解決すべき課題も残っている。
4. 水素原子の化学結合の計測
電子を一つしか持たない水素原子では、結合相手の炭素原子や窒素原子の方に引き寄せられ、X線回折から得られる電子密度マップではピーク位置がシフトすることが知られる。一方、電子回折から得られるクーロンポテンシャルマップでは、入射電子は電子と核の双方の電荷に影響を受けるが、後者の寄与が優勢になる。ピーク位置は中性子回折で観察される核密度に近くなるが同一ではない。ポアソン方程式に基づく理論的な計算では、クーロンポテンシャルマップのピークの位置は核よりやや長くなる。さらに、化学結合の種類によって、水素原子と結合相手の原子との距離は異なる。電子への引力が増す極性の高い結合では、結合距離が短くなる。
今回の実測で得られたXFELの電子密度マップと電子線のクーロンポテンシャルマップで、上記の水素原子からの信号のふるまいを測定できる正確度と精度を示す結果となった(図3(b), (c)及び文献[8]の表2、図4)。メチル及びメチレンのC–H結合に比べ、芳香族のC–H結合ではX線と電子線の違いが、座標の標準不確かさに対してより有意に観測できた。これらの密度の特徴は基本的にC–H結合の極性から生じるものであるが、同時に結合の柔軟性の違いを反映している。すなわち、芳香族のC(sp2)–H結合での水素原子の動きは、他のC(sp3)–H結合よりも結合方向に制限されており、これが水素の視認性とピーク位置の精度の向上に繋がっていると考えられる(文献[8]の図2(d)、表2、図4)。アミドのN(sp2)–H結合では極性が強く、水素のピーク位置の変位はより大きな差となって表れた(文献[8]の表2、図4)。
5. 電荷の計測
イオンの電子散乱曲線を反映し、電子線は電荷に対して高い感度を持つことが予想される[3, 12, 13][3] Yonekura, K., Kato, K., Ogasawara, M., Tomita, M. and Toyoshima, C.: Proc. Natl. Acad. Sci. U. S. A. 112 (2015) 3368-3373.
[12] Yonekura, K. and Maki-Yonekura, S.: J. Appl. Crystallogr. 49 (2016) 1517-1523.
[13] Yonekura, K. et al.: IUCrJ 5 (2018) 348-353.。イオン結合した塩素原子(図3、5では示していないが、文献[8]の図2などを参照)とその周囲の水素原子に部分電荷(図5のH15、H16)を与え計算したR値の変化を調べることで、XFELと比べ、電荷に対して高い感度を持つことが実際に示された。また、部分電荷の値も推定できた(図5)[8][8] Takaba, K. et al.: Nat. Chem. 15 (2023) 491-497.。さらに、クーロンポテンシャルを電子密度に変換して計算した差マップから、各原子の電子の分布を可視化できる可能性も示された(図4(e))。
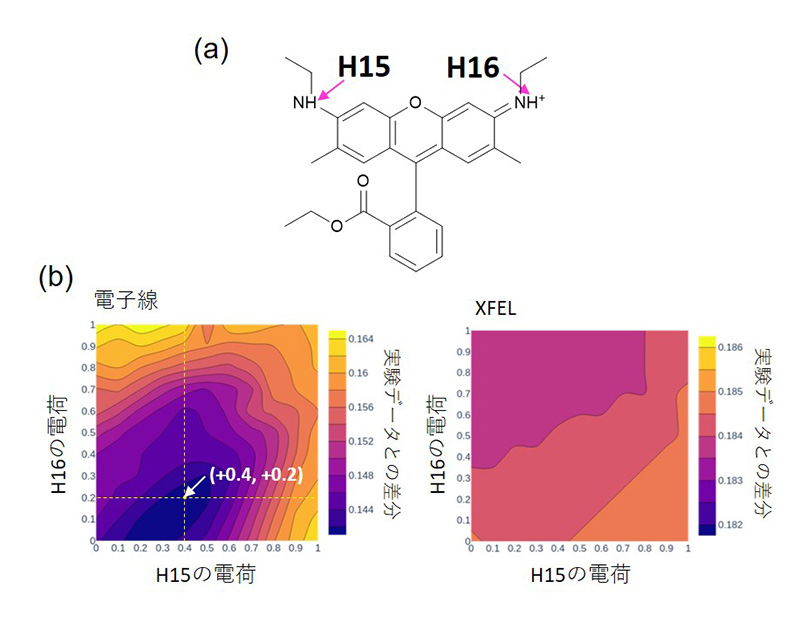
図5 電子回折、X線回折に基づいた最適電荷値の探索。
(a) 正電荷をとりうる水素原子は二つあるが(H15、H16)、一般的な表記では片方の水素に正電荷が付記される。(b) 原子モデル中の二つの水素原子の電荷を縦横の軸に沿って変化させた計算データと実験データの間でR値を計算し、電荷値の組み合わせを探索した。水素の正イオンの散乱因子は文献[13]を参照した。電子線(左)では実験データを最もよく説明する最小のR値(グラフの色の濃い部分)が得られ、二つの水素の両方が部分的に正電荷を帯びていることが実験的に示された。一方でXFEL(右)では電荷を変えても変化が小さく、電荷は決定できない。文献[8]より改変。
6. おわりに
紹介した研究により、XFELを用いて低分子有機化合物の微小な結晶から、その詳細な構造を解析することが可能になった。実証実験として示したローダミン6Gの解析では、微小結晶を分散させた試料支持板を二次元スキャンすることで、XFEL回折像を多数の結晶から集め、同じ試料の電子回折の回転撮影から求めた格子情報を使って指数付けする手順を確立した。これにより、水素原子が可視化できると共に、電子回折のデータに比べ優れた統計値と高い信頼性を持つ原子座標が得られることが分かった。両線源の構造比較では、両者が水素原子の位置を化学結合の種類に依存して正確に区別でき、両構造間での測定距離の違いは、X線と電子の散乱の特性を反映することも明らかになった。また、電子線は電荷に対して高い感度を持つことを実験的に示せた。
これまで、高分解能X線回折、中性子回折、及びその組み合わせは、対象分子内の水素の結合特性やポテンシャル分布など、非水素原子の原子座標を超える情報の取得に利用されてきた。しかし、中性子回折では、mmサイズの非常に大きな結晶が必要であるという制限に変わりはない。XFELと電子回折では、小さい結晶が使用でき測定に試料の特別な処理も必要ないため、より実用的であろう。一方、同じ結晶試料でも、上述のように透過能の違いから、XFELでは厚い結晶、電子回折では薄いものが適している。
開発した技術は、合成有機化学から創薬、材料科学に至る広範な分野で正確かつ高精度な構造解析を実施するために利用できる。さらに、機能性の有機低分子の時間分解解析などへの応用の可能性も広がると期待される。
謝辞
SACLAでの実験は、理研放射光科学研究センターの井上伊知郎研究員、高輝度光科学研究センターの登野健介チームリーダーらの協力の下で行いました。この研究にご協力いただいた皆様、理研の生体機構研究グループの当時のメンバーに感謝申し上げます。
紹介した研究は、日本学術振興会(JSPS)科学研究費助成事業若手研究「電子線を利用したタンパク質の分子内電荷の決定(研究代表者:高場圭章、20K15764)」、科学技術振興機構(JST)未来社会創造事業探索加速型「微小結晶構造の自動・高精度電子線解析(研究代表者:米倉功治、JPMJMI20G5)」、同戦略的創造推進事業CREST「実験・計算・データ科学融合による塗布型電子材料の開発(JPMJCR18J2)」などの助成を受けて行いました。
参考文献
[1] Henderson, R.: Q. Rev. Biophys. 28 (1995) 171-193.
[2] Nannenga, B. L., Shi, D., Leslie, A. G. W. and Gonen, T.: Nat. Methods 11 (2014) 927-930.
[3] Yonekura, K., Kato, K., Ogasawara, M., Tomita, M. and Toyoshima, C.: Proc. Natl. Acad. Sci. U. S. A. 112 (2015) 3368-3373.
[4] Barty, A. et al.: Nat. Photonics 6 (2012) 35-40.
[5] Chapman, H. N., et al.: Nature 470 (2011) 73-78.
[6] Boutet, S. et al.: Science 337 (2012) 362-364.
[7] Schriber, E. A. et al.: Nature 601 (2022) 360-365.
[8] Takaba, K. et al.: Nat. Chem. 15 (2023) 491-497.
[9] Yonekura, K., Ishikawa, T. and Maki-Yonekura, S.: J. Struct. Biol. 206 (2019) 243-253.
[10] Takaba, K., Maki-Yonekura, S., Inoue, S., Hasegawa, T. and Yonekura, K.: Front. Mol. Biosci. 7 (2021) 612226.
[11] Clabbers, M. T. B., Gruene, T., van Genderen, E. and Abrahams, J. P.: Acta Crystallogr. A 75 (2019) 82-93.
[12] Yonekura, K. and Maki-Yonekura, S.: J. Appl. Crystallogr. 49 (2016) 1517-1523.
[13] Yonekura, K. et al.: IUCrJ 5 (2018) 348-353.
(国)理化学研究所 放射光科学研究センター
生体機構研究グループ
東北大学 多元物質科学研究所
〒679-5198 兵庫県佐用郡佐用町光都1-1-1
TEL : 0791-58-2837
e-mail : yone@spring8.or.jp
(国)理化学研究所 放射光科学研究センター
生体機構研究グループ
〒679-5198 兵庫県佐用郡佐用町光都1-1-1
TEL : 0791-58-2837
e-mail : takaba@spring8.or.jp
(国)理化学研究所 放射光科学研究センター
イメージング開発チーム
〒679-5198 兵庫県佐用郡佐用町光都1-1-1
TEL : 0791-58-2837
e-mail : makis@spring8.or.jp




