



Volume 26, No.3 Pages 256 - 260
1. 最近の研究から/FROM LATEST RESEARCH
長期利用課題報告
ゼオライトの精密設計を目的とした非周期系原料及び結晶ゼオライトの原子・ナノスケールPDF解析
Time Resolved Analysis of Zeolite Formation Mechanism at Atomic and Nano Scales: Towards the Design of Novel Functional Zeolites
[1]東京大学 大学院工学系研究科 Department of Chemical System Engineering, The University of Tokyo、[2](公財)高輝度光科学研究センター 放射光利用研究基盤センター 回折・散乱推進室 Diffraction and Scattering Division, Center for Synchrotron Radiation Research, JASRI
- Abstract
- 工業的に重要な材料であるゼオライトはその生成メカニズムが未だ完全には明らかになっていない。本研究ではゼオライトの生成過程を原子・ナノスケールで調べることにより、構造規定剤や構成元素の役割を明確にすることを目指した。様々な系におけるゼオライト前駆体構造の形成過程を解明することに成功した。本稿では得られた最新の結果に関して報告する。
1. はじめに
ゼオライトは持続的社会の形成のために大きく貢献するキーマテリアルといっても過言ではなく、今日では年間100万トン以上製造されており、その市場はゼオライト粉末だけで4,000億円以上ある。さらに、ゼオライトの特性が触媒プロセス、工業プラントの性能・サイズ等を決定しており、その波及効果は極めて大きい。ゼオライトは主に構造規定剤含有アルミノシリケート非晶質を水熱条件下での結晶化により得られるが、出発物質が非晶質であることから、その生成過程は十分に解明されておらず、経験則に基づいたトライアルアンドエラー的なアプローチにより新規材料合成が試みられてきた。今後、こういった材料開発において日本が世界を先導する立場をとるためには、その生成過程を原子・ナノスケールで調べることにより、構造規定剤や構成元素の役割を明確にし、得られた情報に基づいた設計を試みる必要がある。そのためには、回折パターンのブラッグピークの有無にかかわらず、原子・ナノスケールにおける構造情報が直接観測できる、二体分布関数(PDF)解析及び、その情報に基づいた構造モデリングによる3次元構造解析を時分割で行う必要がある。SPring-8の特徴である60 keV以上の高エネルギーX線を用いれば、世界最高レベルのPDFデータを取得することができるため、これを最大限に生かした長期利用課題(2018A0155~2019B0155)を行うこととなった。なお、本長期利用課題は長期利用課題2015A0115~2017B0115から連続して採択された。
2. 研究の意義
ゼオライトはミクロ孔領域(直径 < 2 nm)の大きさの空間を有する結晶であり、シリコンを主としてアルミニウムに代表されるヘテロ原子が酸素原子を介して結合し、構成されている(図1参照)[1][1] T. Wakihara, T. Okubo: Chemistry Letters 34 (2005) 276-281.。
また骨格の負電荷を補償するため、アルカリ金属等のカチオンが含まれる。1950年代にはゼオライト合成が可能となり、今日では年間100万トン以上製造されている。例えば、FAU型ゼオライトは原油中の重質成分をガソリンやナフサに転換するFCC触媒の主要成分である。また、*BEA型、CHA型ゼオライトはディーゼル車のNOx排出規制に対応する触媒として実用化され始めた。以上のように、ゼオライトは持続的社会の形成のために大きく貢献するキーマテリアルである。
ゼオライトが実用に供されるためには、あるニーズに対して構造、形態、組成の3条件が同時最適化されている必要がある。構造に関しては、過去20年間のゼオライト合成技術の進展は目覚ましく、現在では240種類程度の骨格構造が見出されている。現在においても年間5~10程度の新規構造が報告されている。形態に関しては、ゼオライトの核発生・結晶成長を制御するボトムアップ手法、もしくは筆者が開発した粉砕と再結晶化法を組み合わせるトップダウン法により、大半のニーズに対応できるようになった[2][2] T. Wakihara, R. Ichikawa, J. Tatami, A. Endo, K. Yoshida, Y. Sasaki, K. Komeya and T. Meguro: Crystal Growth & Design 11 (2011) 955-958.。一方、組成に関してはその自由度が極めて制限されているのが現状である。ゼオライトを触媒として利用する際、ゼオライト骨格中の活性点(= Al)が多過ぎるとコーキングが促進されてしまうため、適度なSi/Al組成(4~50)を持つことが求められる。しかし、Si/Al比が1~2近傍しか合成できない構造、もしくはピュアシリカタイプしか合成できない構造が多く、結果として実用に供されていないゼオライトが多くある。これは、一般的に速度論的な影響を強く受ける一方、これらの要素を合理的に制御する手段が限られているためである。以上より、これまでに報告例のない組成を持つゼオライト合成には大きなハードルとともにニーズがあり、従来限界を超える組成の新規性を持つゼオライトの設計のため、新しい合成手法の概念をゼオライトサイエンスに導入する必要がある。本研究ではPDFを始めとした分析手法を駆使し、新しい合成につながる知見を得ることを試みた結果の一部を報告する。

図1 代表的なゼオライト種。
3. 成果の一例
3-1. ゼオライト結晶化メカニズムの時分割解析[3][3] H. Yamada, S. Tominaka, K. Ohara, Z. Liu, T. Okubo and T. Wakihara: The Journal of Physical Chemistry C 46 (2019) 28419-28426.
これまでの研究ではex situ条件下での解析が主であり、ゼオライト合成過程の原子レベルでの直接観察は困難であった。そのため本章ではゼオライトの合成過程においてin situ条件下で高エネルギーX線全散乱(High-Energy X-ray Total Scattering: HEXTS)法を適用し、ゼオライト前駆体の構造形成過程を原子レベルで直接観察することを目指した。まずゼオライト合成過程をin situ条件下で観察可能なビームラインを立ち上げ、その後ゼオライト合成過程のその場分析を実現した。研究対象としては工業的に広く用いられているゼオライトであるMFI型及び*BEA型とした。
ゼオライト合成過程のin situ HEXTS実験は以下の手順で実施した。SPring-8のBL08Wにフラットパネルディテクタ及びゼオライト合成に使用するマイクロ波合成装置を設置し、迷光等を除去可能な光学系の調整を行った。使用したエネルギーは115 keVであり、0.50−20 Å-1までのQ領域のデータを1分ごとに測定を実施した。さらに、得られたデータに対し合成開始後の散乱パターンからの差分を算出してrelative PDFパターンを算出し、合成開始時点からの変化分のみを抽出した。MFI型及び*BEA型ゼオライトの合成は既報を参考に実施した。
MFI型及び*BEA型ゼオライトのin-situ PDFパターンを図2a)、b)に示す。これらの時分割PDFパターンから、両ゼオライトの結晶化過程において、合成の初期の段階において最終生成物と類似した近距離構造が既に形成していることが確認された。しかしながらゼオライトの核生成が生じ結晶成長の段階に入ると、短距離側にハイライトされた相関が両ゼオライトで徐々に増加していく一方、誘導時間中においてはその構造変化は小さく全散乱データから前駆体の構造形成を観察することは困難であった。
Relative PDFを図2c)、d)に示す。誘導期間中に徐々にアルミノシリケート種の重合が進行した影響で1.6、2.6、3.1 Åの相関がそれぞれ減少、増加、増加したと考えられる。また、長距離領域でハイライトされた領域に着目すると、これらの相関が徐々に短距離側が減少し、長距離側の相関が増加していく傾向が確認された。これは、結晶化前の誘導期間中に溶解・再析出を経て段々と大きなリング構造体が形成してくることを示唆している。以上のように、本手法を用いることで、誘導期間中においてアルミノシリケート種の縮合が進行し、ゼオライト前駆体中のリング分布が徐々に変化していく過程が初めて明らかになった。
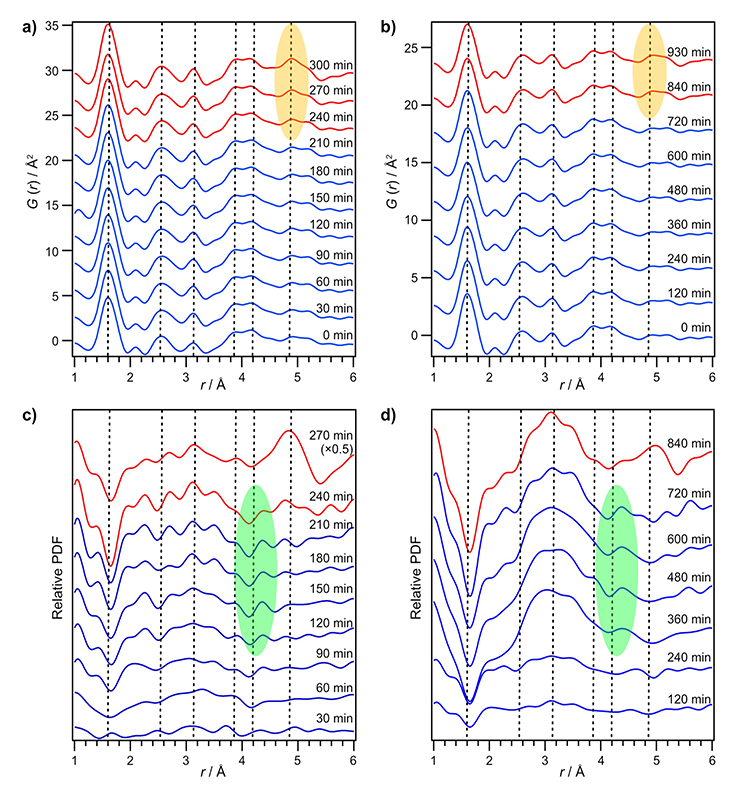
図2 In situ条件下におけるPDF及びrelative PDF。a) MFI型ゼオライトのPDF、b) *BEA型ゼオライトのPDF、c) MFI型ゼオライトのrelative PDF、d) *BEA型ゼオライトのrelative PDF。
3-2. 合成途中の攪拌処理によるkinetics制御[4][4] C.-T. Chen, K. Iyoki, H. Yamada, S. Sukenaga, M. Ando, H. Shibata, K. Ohara, T. Wakihara and T. Okubo: The Journal of Physical Chemistry C 33 (2019) 20304-20313.
ハイドロゲルからのゼオライトの結晶化は、通常長い合成期間を必要とし、その結果幅広い粒度分布を持つ多結晶の凝集体の形で生成物が得られる。本研究では140°Cで有機物を含まないハイドロゲル(0.275 Na2O: 0.025 Al2O3: 1 SiO2: 25 H2O)からモルデナイト(MOR)を合成した。なお、合成の途中で反応器を急冷し、ヘラを用いて手で1分間撹拌する「中間撹拌法」により沈降したバルク固相を分解する作業を行った。
静置した状態で140時間合成した場合においても完全な結晶性の製品を得るには不十分である。しかし、合成系を40時間撹拌した場合、同じ合成時間内に完全に結晶化させることができた。反応器を冷却したり開放したりしても、この系の結晶化速度には影響しないことが対照実験で確認された。したがって、中間撹拌プロセスが本ゼオライトの結晶化に寄与したと考えられる。
さらに、中間攪拌を行うタイミングも、攪拌後の系の結晶化速度に影響を与えることが分かった。例えば、90時間及び110時間の時点で撹拌を実施した場合、その後のモルデナイトの結晶化が短時間で完了することが確認された。また、結晶生成物の粒子径や粒度分布(PSD)は、撹拌タイミングによって変化した。例えば、20時間の撹拌ではPSDの広い大きな結晶(約13 μm)が得られたが、90時間、110時間の撹拌ではPSDの狭い小さな結晶(約4 μm)が形成された。これらのサンプルに対し、NMR及びPDFを用いてアルミノシリケートリングの形成過程や重合状態の中間攪拌前後での変化を詳細に解析した結果(図3)、本システムにおけるモルデナイトの結晶化は自己触媒的な核生成挙動を示すことが推測された。これは、固相に埋め込まれた核が、非晶質粒子の溶解に伴って液相と接触することで初めて成長するというモデルである。つまり、モルデナイトの核の数は合成時間とともに徐々に増えていくものの、アルミノシリケートのマトリックスが凝縮しているため、核の成長が大きく妨げられていた状態にとどまっていた。そこに中間攪拌を行うことで、バルク状の固体が小さな断片になり固相と液相の間の化学種の交換が促進されるため、これらの埋め込まれた核の成長が活性化されたものと考えられる。
以上のように、中間攪拌法を用いて高密度ハイドロゲル系におけるモルデナイトの核生成挙動を明らかにし、固相と液相が効果的に接触することでアルミノシリケートの再形成、特に結晶成長が促進されることを示した。
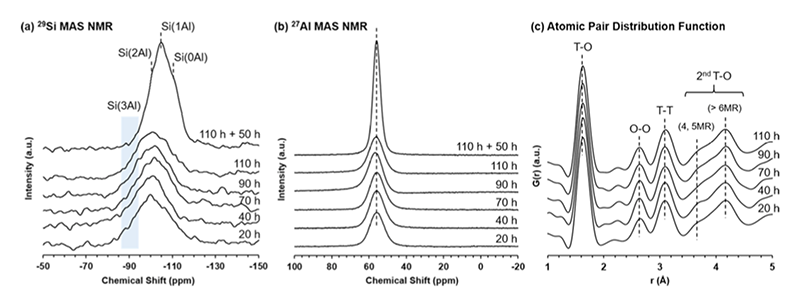
図3 MOR型ゼオライト合成過程におけるNMR及びPDF解析結果。
3-3. 原料初期構造がゼオライト結晶化に与える影響の解明[5][5] C.-T. Chen, K. Iyoki, P. Hu, H. Yamada, K. Ohara, S. Sukenaga, M. Ando, H. Shibata, T. Okubo and T. Wakihara: Journal of the American Chemical Society just accepted.
反応物混合物の調製や水熱合成の際に生成されるアルミノシリケート種の構造は、ゼオライトの結晶化kineticsに重要な役割を果たす。異なる反応物混合物調製法により、80°Cで高密度ハイドロゲル系(1 Na2O: 0.2 Al2O3: 1 SiO2: 50 H2O)からゼオライトX(FAU)を合成した(図4)。Cab-O-Sil系では、最後のステップとしてAl含有溶液にCab-O-Silを添加した。溶解Cab-O-Sil系では、まずCab-O-SilをNaOH(aq)に溶解し、次いでAl含有溶液を添加した。
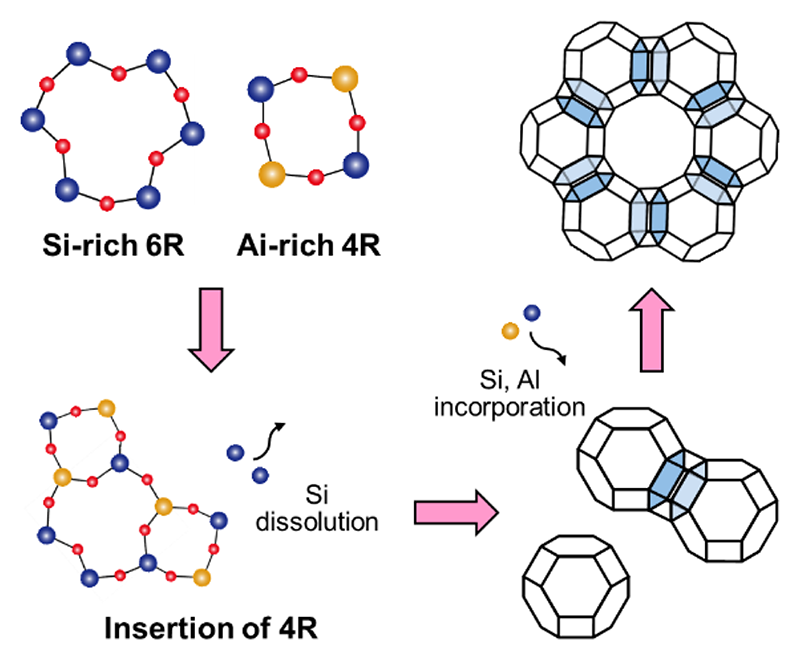
図4 本システムのゼオライトXの生成スキーム。
Cab-O-Sil系でのゼオライトXの結晶化は、溶解したCab-O-Sil系での結晶化よりもはるかに速く、(前者の系では後者の系(約15 μm)に比べて小さな結晶(約1.2 μm)が生成された。この結果は、Cab-O-Sil系での急速な結晶化が、不均一核生成によるものであることを示している。またハローピークの位置の違いが示唆しているように、2つのシステムにおけるアモルファス物質の構造とその形成過程はおそらく異なると推察される。また、組成分析の結果、Cab-O-Sil系のSi種が誘導期間中に液相に放出され、結晶成長中に固相に取り込まれることを示しているが、一方で、溶解したCab-O-Sil系の液相中のSi種の濃度はほぼ一定であったことが明らかになった。
さらに、アモルファス物質の構造変化を明らかにするため、いくつかの特性評価法を用いて固体生成物を分析した。ラマン分光法により、両系の非晶質アルミノシリケートは大部分が4-MRで構成されていることが明らかになった。注目すべきは、これらの4-MRバンドが結晶成長期間中に著しく強まっていることであり、これは4-MRの存在量が増加していることを意味する。この現象は、D6Rが既存の4-MRの環化によって生じたのではなく、2つの6-MRの組み合わせによって形成されたことを示しており、この過程でさらに6つの4-MRが生成されたことになる。興味深いことに、溶解したCab-O-Silシステムでの44時間のサンプルのD6Rバンドは、Cab-O-Silシステムでの6時間のサンプルと比較して、ほとんど識別できなかった。両サンプルの結晶化度が約50%と類似していることから、Cab-O-Silシステム中の非晶質物質はD6Rを中心とした部分的に秩序のある構造を既に有しているが、溶解したCab-O-Silシステム中にそのような周期構造が存在しないことは妥当と考えられる。
固体核磁気共鳴(NMR)分光法により、Cab-O-Sil系のAl原子は、加熱により比較的小さいSi-O-Al結合角を持つ構造に速やかに取り込まれ、この状態は合成中も変化しないことが確認された。また、放射光X線全散乱データから得られたアモルファス物質のPDFの結果から、Cab-O-Silシステムの非晶質中には、合成後1時間でSiに富む6-MRとAlに富む4-MRが既に形成されていることが分かった。さらに加熱すると、6-MRの原子対間距離が拡大したが、これはAl原子が6-MRに取り込まれたことによるものである。これらの結果から、(1)誘導期間中に非晶質中のSi量が徐々に減少し(元素分析)、(2)構造組成(XRD)及びAl原子の位置(NMR)は変化せず、(3)6-MRが徐々にAlリッチになり(PDF)、(4)6-MR間の結合によりD6Rが形成されたことから、Cab-O-Sil系ゼオライトXの結晶化メカニズムが提案された(図4)。
一方、溶解したCab-O-SilをSi源として用いた場合、合成開始後すぐに歪んだ構造と大きな環を持つアルミノシリケートが形成された。これは、2つの最近接O原子間の距離が大きく(PDF)、Si-O-Al結合の角度が大きい(NMR)ことから示唆された。このアルミノシリケート構造は、80°Cの加熱により大きく歪んだものとなった。その結果、これらのアルミノシリケートを再編成して長距離秩序構造を作り出すことは容易ではなく、より長い合成時間を必要とする。
なお、PDF解析の結果から、混合物を室温でエージングさせると、アルミノシリケートのリング構造の歪みの度合いが小さくなることが分かった。RTで24時間エージングさせた後、80°Cで24時間加熱すると、約3.6 μmのゼオライトXの結晶が得られた。つまり、総合成時間は48時間であった。非エージング系では80°Cで50時間以上の合成が必要であったことを考えると、歪んだアルミノシリケート構造は高温で安定化すると考えられる。
4. まとめ
ゼオライトの生成過程を原子・ナノスケールで調べることにより、構造規定剤や構成元素の役割を明確にすることを目指した。様々な系におけるゼオライト前駆体構造の形成過程を解明することに成功した。さらに、時分割測定に関してもその測定を実現し、非晶質ゼオライト前駆体中の構造形成を明らかにする礎を築いた。今後はこれらの技術を組み合わせて様々なゼオライト合成系にこれらの知見を展開していく予定である。
参考文献
[1] T. Wakihara, T. Okubo: Chemistry Letters 34 (2005) 276-281.
[2] T. Wakihara, R. Ichikawa, J. Tatami, A. Endo, K. Yoshida, Y. Sasaki, K. Komeya and T. Meguro: Crystal Growth & Design 11 (2011) 955-958.
[3] H. Yamada, S. Tominaka, K. Ohara, Z. Liu, T. Okubo and T. Wakihara: The Journal of Physical Chemistry C 46 (2019) 28419-28426.
[4] C.-T. Chen, K. Iyoki, H. Yamada, S. Sukenaga, M. Ando, H. Shibata, K. Ohara, T. Wakihara and T. Okubo: The Journal of Physical Chemistry C 33 (2019) 20304-20313.
[5] C.-T. Chen, K. Iyoki, P. Hu, H. Yamada, K. Ohara, S. Sukenaga, M. Ando, H. Shibata, T. Okubo and T. Wakihara: Journal of the American Chemical Society just accepted.
東京大学 大学院工学系研究科
〒113-8656 東京都文京区本郷7-3-1
TEL : 03-5841-7368
e-mail : wakihara@chemsys.t.u-tokyo.ac.jp
東京大学 大学院工学系研究科
〒113-8656 東京都文京区本郷7-3-1
TEL : 03-5841-7368
(公財)高輝度光科学研究センター
放射光利用研究基盤センター 回折・散乱推進室
〒679-5198 兵庫県佐用郡佐用町光都1-1-1
TEL : 0791-58-0833
e-mail : h_yamada@spring8.or.jp




