



Volume 24, No.4 Pages 376 - 382
1. 最近の研究から/FROM LATEST RESEARCH
Long-term Proposal Report 1
NRVS of Mononuclear and Binuclear Non-heme Iron Enzyme Intermediates and Related Model Complexes
[1]Department of Chemistry, Stanford University、[2]SLAC National Accelerator Laboratory、[3]Korea Advanced Institute of Science and Technology (KAIST)
- Abstract
- Non-heme iron (NHFe) enzymes catalyze a wide range of reactions important in both health and technological development. These enzymes activate dioxygen to form transient intermediates capable of performing difficult chemical reactions. Understanding the geometric and electronic structures of these intermediates is essential in elucidating the mechanism of NHFe enzyme catalysis. We have developed an experimental and computational approach using nuclear resonance vibrational spectroscopy (NRVS) to define the structures of the intermediates and to evaluate the enzymatic reaction coordinates. Herein, we report our recent advances using NRVS to understand the importance of Fe(III)-superoxide intermediates in mononuclear NHFe enzymes, as well as the unique ability of binuclear NHFe enzymes to react with substrates on the Fe(III)2-OOH level.
Introduction
Non-heme iron (NHFe) enzymes are ubiquitous in Nature and catalyze key reactions in human health, metabolism, and bioremediation, among many other functions. The prominent role of these enzymes in all realms of life have made them targets for breakthroughs in chemistry and biology. Generally, these enzymes use one or two ferrous ions to react with molecular oxygen in forming a substrate-reactive intermediate and are subdivided into classes based on the types of intermediates formed and substrates utilized (Figure 1). The chemistry performed by these enzyme intermediates spans broadly and includes hydrogen-atom abstraction (HAA), ring closure and expansion, electrophilic aromatic substitution (EAS), hydroxylation and halogenation[1][1] Solomon, E. I.; Brunold, T. C.; Davis, M. I.; Kemsley, J. N.; Lee, S. K.; Lehnert, N.; Neese, F.; Skulan, A. J.; Yang, Y. S. and Zhou, J.: Chem. Rev. 2000, 100, 235-350.. Thus, a longstanding goal has been to determine the geometric and electronic structure of NHFe enzyme intermediates, and to correlate their unique structures with chemical function. To this end, our lab has developed a combined experimental and computational methodology using nuclear resonance vibrational spectroscopy (NRVS) and density functional theory (DFT). NRVS probes the vibrational sidebands of the iron Mossbauer transition with spectral intensity proportional to iron displacement in a vibration; hence NRVS is site-selective for the catalytic center and yields rich spectroscopic information relating to the iron coordination environment. We collect NRVS spectra on freeze-quench trapped oxygen intermediates and simulate these data with DFT calculations to illuminate their geometric/electronic structures and evaluate their reaction coordinates in catalysis. This technique has led to major findings in the mononuclear NHFe family of enzymes. Our previous NRVS study on the alpha ketoglutarate-dependent (αKG) enzyme syringomycin halogenase (SyrB2) defined the high-valent Fe(IV)=O intermediate responsible for L-threonine halogenation[2][2] Wong, S. D.; Srnec, M.; Matthews, M. L.; Liu, L. V.; Kwak, Y.; Park, K.; Bell, C. B., III; Alp, E. E.; Zhao, J.; Yoda, Y.; Kitao, S.; Seto, M.; Krebs, C.; Bollinger, J. M., Jr. and Solomon, E. I.: Nature 2013, 499, 320-323., and provided major insight into how orientation of the Fe(IV)=O unit relative to the substrate controls preferential halogenation over the thermodynamically-favored hydroxylation reaction[3,4][3] Srnec, M.; Wong, S. D.; Matthews, M. L.; Krebs, C.; Bollinger, J. M., Jr. and Solomon, E. I.: J. Am. Chem. Soc. 2016, 138, 5110-5122.
[4] Srnec, M. and Solomon, E. I.: J. Am. Chem. Soc. 2017, 139, 2396-2407.. We have also utilized NRVS to define the low-spin hydroperoxide intermediate in activated bleomycin (ABLM), and through DFT have shown how the hydroperoxide is primed for the double-strand cleavage of DNA in anticancer activity[5][5] Liu, L. V.; Bell, C. B., III; Wong, S. D.; Wilson, S. A.; Kwak, Y.; Chow, M. S.; Zhao, J.; Hodgson, K. O.; Hedman, B. and Solomon, E. I.: Proc. Natl. Acad. Sci. 2010, 107, 22419-22424.. We have since applied our experimental and computational methods developed in the SyrB2/ABLM studies to two new classes of mononuclear as well as binuclear NHFe enzymes. In this report, we review our progress in using NRVS to elucidate the intermediates present in the mononuclear Rieske and extradiol dioxygenases (RDO and EDO, respectively), as well as in the binuclear enzyme p-aminobenzoate N-oxygenase (AurF). We conclude by outlining future NRVS studies of NHFe enzymes.
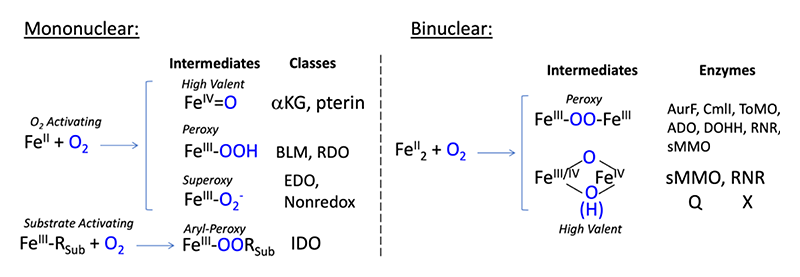
Figure 1 Intermediates of mononuclear (left) and binuclear (right) non-heme enzyme subclasses. Abbreviations: αKG: alpha-keto-glutarate dependent dioxygenase; pterin: pterin-dependent hydroxylase; BLM: bleomycin; RDO: Rieske dioxygenase; EDO: extradiol dioxygenase; Nonredox: non-redox active enzymes; IDO: intradiol dioxygenase. AurF: p-aminobenzoate N-oxygenase. CmlI: arylamine oxygenase of the chloramphenicol pathway; ToMO: toluene o-xylene monooxygenase; ADO: aldehyde-deformylating oxgenase; DOHH: deoxyhypusine hydroxylase; sMMO: soluble methane monooxygenase; RNR: ribonucleotide reductase.
Methodology
NRVS data were collected at SPring-8 BL09XU using C-mode. Raw data were analyzed using the PHOENIX software developed by W. Stuhrhahn[6][6] Sturhahn, W.: Hyperfine Interactions 2000, 125, 149-172. and the online spectra.tools adaptation by L. Gee[7][7] L. B. Gee, spectra.tools: https://www.spectra.tools/. The NRVS energy scales were calibrated with [FeIII(Cl4)]NEt4[8][8] Smith, M. C.; Xiao, Y.; Wang, H.; George, S. J.; Coucouvanis, D.; Koutmos, M.; Sturhahn, W.; Alp, E. E.; Zhao, J. and Cramer, S. P.: Inorg. Chem. 2005, 44, 5562-5570.. DFT calculations were performed using Gaussian 09 and ORCA packages installed on an in-house cluster. DFT functionals and basis sets were chosen from calibrated studies on well-defined model complexes; for details, see references[9,10][9] Park, K.; Tsugawa, T.; Furutachi, H.; Kwak, Y.; Liu, L. V.; Wong, S. D.; Yoda, Y.; Kobayashi, Y.; Saito, M.; Kurokuzu, M.; Seto, M.; Suzuki, M. and Solomon, E. I.: Angewandte Chemie Int-Ed 2013, 52, 1294-1298.
[10] Park, K.; Bell, C. B.; Liu, L. V.; Wang, D.; Xue, G.; Kwak, Y.; Wong, S. D.; Light, K. M.; Zhao, J.; Alp, E. E.; Yoda, Y.; Saito, M.; Kobayashi, Y.; Ohta, T.; Seto, M.; Que, L. and Solomon, E. I.: Proc. Natl. Acad. Sci. 2013, 110, 6275-6280. (doi: 10.1073/pnas.1304238110). Models for intermediates were chosen from related crystal structures or available EXAFS data. Truncation schemes for the DFT models has been described[11][11] Park, K. and Solomon, E. I.: Can. J. Chem. 2014, 92, 975-978.; briefly, amino acids are truncated at the Cα with two hydrogen atoms, resulting in all amino acid side chains initiating with a methyl group. For vibrational simulations, the mass of the two substituted hydrogens was increased to 100 to prevent artificial vibrational mixing of the methyl group into iron modes.
Results
Fe(III)-peroxy Intermediate in the Benzoate 2,3-Dioxygenase Peroxide Shunt Reaction
In our previous report, our NRVS studies on Fe-peroxy intermediates elucidated the active intermediate in the glycopeptide anticancer drug bleomycin, a low-spin Fe(III)-hydroperoxide responsible for DNA cleavage[12][12] Liu, L. V.; Bell, C. B.; Wong, S. D.; Wilson, S. A.; Kwak, Y.; Chow, M. S.; Zhao, J.; Hodgson, K. O.; Hedman, B. and Solomon, E. I.: Proc. Natl. Acad. Sci. 2010, 107, 22419-22424.. Insight into the structure of this intermediate allowed evaluation of the reaction coordinate, establishing key differences in the reactivity of heme vs non-heme Fe(III)-peroxy intermediates[13][13] Decker, A.; Chow, M. S.; Kemsley, J. N.; Lehnert, N. and Solomon, E. I.: J. Am. Chem. Soc. 2006, 128, 4719-4733.. Iron-peroxy intermediates have also been observed in the Reiske and extradiol dioxygenase NHFe enzyme classes, however, these intermediates are high-spin thus motivating our NRVS investigation of two high-spin Fe(III)-(hydro)peroxy models where we defined key spectroscopic handles for distinguishing the influence of peroxide binding mode and protonation state[14][14] Sutherlin, K. D.; Liu, L. V.; Lee, Y. M.; Kwak, Y.; Yoda, Y.; Saito, M.; Kurokuzu, M.; Kobayashi, Y.; Seto, M.; Que, L. Jr.; Nam, W. and Solomon, E. I.: J. Am. Chem. Soc. 2016, 138, 14294-14302.. During this long-term proposal period, we have greatly enhanced our understanding of Fe(III)-peroxy intermediates by assigning and characterizing the intermediate in the peroxide shunt reaction of the Rieske dioxygenase, benzoate 1,2-dixoygenase (BZDO).
BZDO catalyzes the cis-dihydroxylation of benzoate and like other RDOs is an important target for enzyme derived bioremediation catalysts[1][1] Solomon, E. I.; Brunold, T. C.; Davis, M. I.; Kemsley, J. N.; Lee, S. K.; Lehnert, N.; Neese, F.; Skulan, A. J.; Yang, Y. S. and Zhou, J.: Chem. Rev. 2000, 100, 235-350.. We have trapped and characterized, BZDOp, the intermediate in the peroxide shunt reaction shown to be capable of product formation. Figure 2 shows the NRVS data of BZDOp and key controls in the assignment of its geometric structure. All RDOs contain an iron-sulfur (Rieske) cluster and a catalytic NHFe center. Thus to distinguish the features arising from the Rieske center we compared NRVS on apo-BZDO (NHFe center removed, Rieske center intact) and BZDO (both NHFe and Rieske center intact), Figure 2 black and red traces respectively. We further obtained high quality H216/18O2 isotope data on BZDOp observing an isotope dependent feature (the νFe-OO(H) mode) near 500 cm-1, Figure 2 blue trace and inset. Using DFT simulations and comparing to our high-spin Fe(III)-(hydro)peroxy model complex NRVS study we assigned BDZOp binding mode and protonation state to be a side-on Fe(III)-hydroperoxy species.
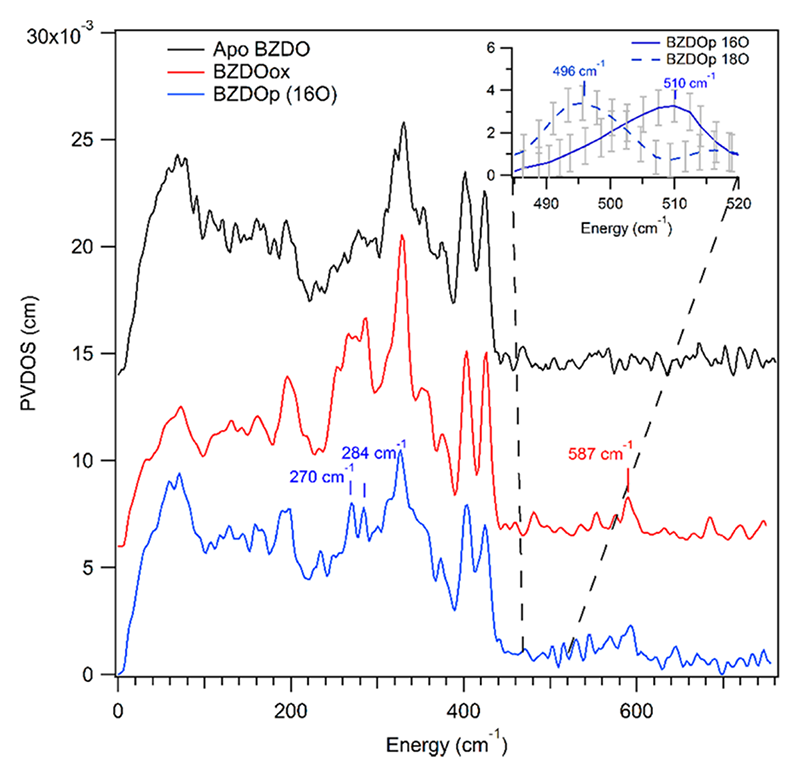
Figure 2 BZDO NRVS data on apo-BZDO, Rieske site intact, catalytic NHFe site depleted (black trace), BZDOox, both Rieske and NHFe site intact (red trace), BZDOp, peroxide shunt intermediate generated with H216O2 (blue trace). Inset: Focus region scans with high S/N depicting H216O2 and H218O2 isotopic dependent feature near 500 cm-1.
With BZDOp structure defined, we extended our study to the peroxide shunt and native O2-dependent reaction coordinate calculations. In the peroxide shunt reaction, the most viable pathway proceeded through heterolytic cleavage of the peroxide bond to form a high valent Fe(V) intermediate capable of cis-dihydroxylation via a large barrier consistent with the slow experimental kinetics of the shunt reaction. In contrast, the native O2 reaction calculations revealed an Fe(III)-superoxo intermediate capable of electrophilic attack on benzoate with a low barrier consistent with rapid O2 reactivity. While the binding of the O2 to Fe(II) to form the Fe(III)-superoxo is endergonic, this reaction is driven by the PCET from the Rieske center. Importantly, our NRVS/DFT methodology showed that the BZDO peroxide shunt and native dioxygen reactions occur via different mechanisms both capable of producing the cis-dihydroxylated product, but with the Fe(III)-O2- being far more reactive[15][15] Sutherlin, K. D.; Rivard, B. S.; Böttger, L. H.; Liu, L. V; Rogers, M. S.; Srnec, M.; Park, K.; Yoda, Y.; Kitao, S.; Kobayashi, Y.; Saito, M.; Seto, M.; Hu, M.; Zhao, J.; Lipscomb, J. D. and Solomon, E. I.: J. Am. Chem. Soc. 2018, 140, 5544-5559..
Intermediates-1 and 2 in Homoprotocatechuate 2,3-Dioxygenase
Extradiol dioxygenases catalyze the aromatic ring cleavage of catechol derived substrates with concomitant incorporation of dioxygen. Like the RDOs, EDOs have generated much interest in the field of microbial bioremediation[1][1] Solomon, E. I.; Brunold, T. C.; Davis, M. I.; Kemsley, J. N.; Lee, S. K.; Lehnert, N.; Neese, F.; Skulan, A. J.; Yang, Y. S. and Zhou, J.: Chem. Rev. 2000, 100, 235-350.. Our NRVS studies on the EDO, homoprotocatechuate 2,3-dioxygenase (HPCD) focused on two trappable solution phase intermediates, Int-1 which decays to Int-2, in the reaction of the H200N-HPCD variant with the slow nitrocatecholate substrate (4NC)[16][16] Groce, S. L. and Lipscomb, J. D.: Biochemistry 2005, 44, 7175-7188.. The electronic structures of Int-1 and Int-2 had been evaluated, however geometric structural O2 binding modes and protonation states were unknown. Figure 3 shows the NRVS data (A and D), the DFT simulations (B and E) and calculated structures (C and F) for Int-1 and Int-2, respectively. Analysis identified the key vibrational modes: ν1 ν-Fe-O2, ν2 δ-Fe-O2, ν3 δ-transaxial, ν4 z-translation and ν5 δ-Fe-substrate. These provided the geometric structures of Int-1 and Int-2 as an end-on Fe(III)-superoxo-catecholate and an end-on Fe(III)-hydroperoxy-semiquione species, respectively.
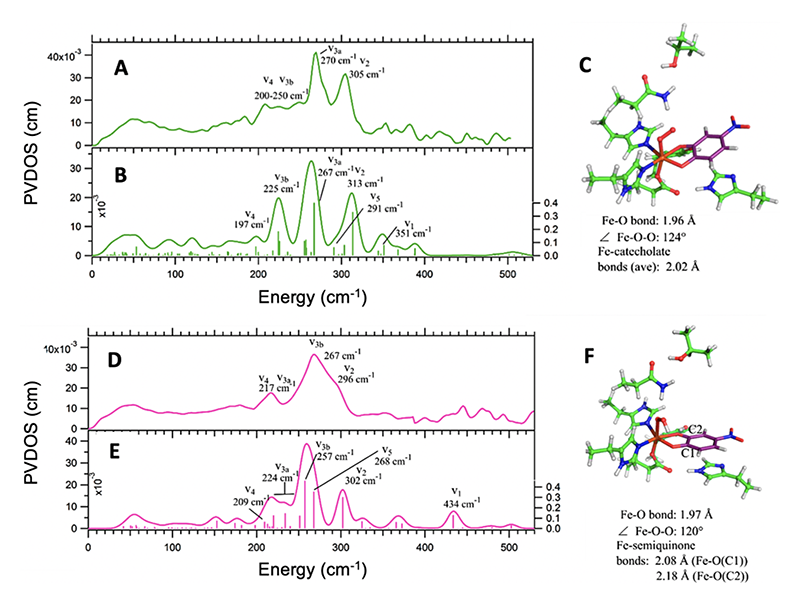
Figure 3 Top: Experimental NRVS data of Int-1 (A) with DFT spectral simulation (B) of structure (C) assigning Int-1 as a Fe(III)-superoxo-catecholate. Bottom: Experimental NRVS data of Int-1 (D) with DFT spectral simulation (E) generated from the end-on Fe(III)-hydroperoxy-semiquinone species (F). Key vibrational modes used in structural assignment are indicated (description in the text).
H200 is proposed to serve as an important second sphere proton acceptor/donor residue in extradiol catalysis, and thus to evaluate its role, we extended our study to DFT calculations of the wt-HPCD-4NC crystallographically characterized intermediates[17][17] Kovaleva, E. G. and Lipscomb, J. D.: Science. 2007, 316, 453-457.. Our key findings from this analysis[18][18] Sutherlin, K. D.; Wasada-Tsutsui, Y.; Mbughuni, M. M.; Rogers, M. S.; Park, K.; Liu, L. V.; Kwak, Y.; Srnec, M.; Böttger, L. H.; Frenette, M.; Yoda, Y.; Kobayashi, Y.; Kurokuzu, M.; Saito, M.; Seto, M.; Hu, M.; Zhao, J.; Alp, E. E.; Lipscomb, J. D. and Solomon, E. I.: J. Am. Chem. Soc. 2018, 140, 16495-16513. were: 1. Fe(III)-(hydro)peroxy intermediates in both H200N and wt-HPCD are precluded from direct attack on substrate by a large activation barrier; 2. While H200N-HPCD-4NC Int-2, the Fe(III)-hydroperoxo, is a dead-end species that does not ring cleave, the presence of H200 in wt-HPCD allows for deprotonation to form an isoenergic Fe(III)-superoxo- catecholate species with a π*in-plane frontier molecular orbital capable of direct attack on the coordinated catechol substrate with a low barrier; 3. Finally, the presence of H200 stabilizes the thermodynamics of Fe-alkylperoxy-bridge formation a key step in extradiol reactivity. Future studies will focus on the understanding the Fe-alkylperoxo-bridge intermediate in wt-HPCD with native substrate, where NRVS data have been collected and analysis is currently underway. This intermediate is proposed to control extra- vs intradiol catecholate ring cleavage and therefore is key in understanding the chemistry of EDOs and the related intradiol dioxygenases.
Together our NRVS studies on RDO and EDO have greatly built upon our understanding of mononuclear Fe(III)-(hydro)peroxy intermediates but importantly have revealed that Fe(III)-superoxy intermediates are in fact the key active species in mononuclear NHFe chemistry. The lack of reactivity of the mononuclear Fe(III)-(hydro)peroxy species provides an important contrast to binuclear systems, in which the binuclear hydroperoxy intermediates are highly reactive as described in our following study on AurF.
Intermediate P' in AurF
Binuclear non-heme iron enzymes (NH2Fe) activate molecular oxygen to perform diverse chemistries, including oxygenation of organic substrates and hydrogen atom abstraction[19][19] Park, K.; Li, N.; Kwak, Y.; Srnec, M.; Bell, C. B.; Liu, L. V.; Wong, S. D.; Yoda, Y.; Kitao, S.; Seto, M.; Hu, M.; Zhao, J.; Krebs, C.; Bollinger, J. M., Jr. and Solomon, E. I.: J. Am. Chem. Soc. 2017, 139, 7062-7070. (doi: 10.1021/jacs.7b02997). The general reaction mechanism for NH2Fe enzymes involves initial activation of O2 to form peroxo intermediate P that converts to the reactive intermediate that directly performs the oxygenation chemistry. These reactive intermediates can be divided into two classes 1) high valent intermediates formed by the four-electron activation of O2 and 2) the two-electron activation of molecular oxygen to form biferric peroxy intermediates termed intermediate P' that is reactive. While the high-valent reactive intermediates have received much attention, the latter class of reactive intermediates had not been structurally characterized until our spectroscopic investigation of AurF. Here we present our completed analysis of its P' intermediate[1][1] Solomon, E. I.; Brunold, T. C.; Davis, M. I.; Kemsley, J. N.; Lee, S. K.; Lehnert, N.; Neese, F.; Skulan, A. J.; Yang, Y. S. and Zhou, J.: Chem. Rev. 2000, 100, 235-350..
NRVS spectroscopy was used to characterize the P' intermediate in AurF and its reactivity using 16O2/18O2 perturbation, summarized in Figure 4a and 4b respectively. The vibrational spectra for both the 16O2 and 18O2 P' intermediate can be divided into three regions. Region 1 consists of energies below 180 cm-1 and the spectra are relatively featureless, region 2 contains four features at 200, 246, 315 and 363 cm-1 and shows no isotope dependent features, and region 3 contains two oxygen isotope sensitive features above 370 cm-1. These features where assigned to the Fe-O stretches associated with the O2-derived bridging ligand[19][19] Park, K.; Li, N.; Kwak, Y.; Srnec, M.; Bell, C. B.; Liu, L. V.; Wong, S. D.; Yoda, Y.; Kitao, S.; Seto, M.; Hu, M.; Zhao, J.; Krebs, C.; Bollinger, J. M., Jr. and Solomon, E. I.: J. Am. Chem. Soc. 2017, 139, 7062-7070. (doi: 10.1021/jacs.7b02997).
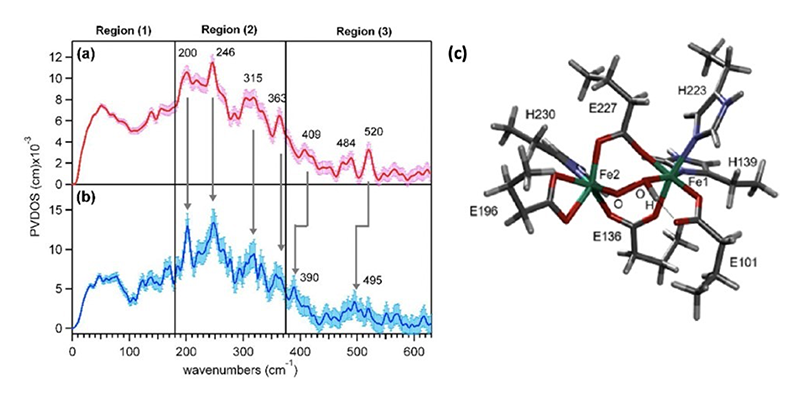
Figure 4 The NRVS spectrum of P' obtained with (a) 16O2 and (b) 18O2. (c) The NRVS derived structure of the P' intermediate in AurF.
To search for possible structures of P', potential intermediates were computationally generated by adding O2 to the DFT optimized structure of biferrous AurF and their NRVS spectra were calculated. Twenty-eight structures were generated with various geometries of the bridging peroxo, different protonation states of the peroxo bridge, as well as differential coordination of a solvent water molecule. Only the model with a µ-1,2-hydroperoxo bridge was able to reproduce the experimental NRVS spectrum, in particular, the large splitting of the oxygen sensitive vibrations at 409 and 520 cm-1 (Figure 4c). This spectroscopically defined hydroperoxy intermediate was then evaluated for its reactivity with the p-aminobenzoate substrate by calculating the potential energy surface (PES) and reaction coordinate (RC), summarized in Figure 5. The oxidation of the substrate by one-electron triggers the rate-limiting cleavage of the O-O hydroperoxo bond and formation of a Fe(IV)=O poised to attack the radical substrate amine. Homolytic cleavage of the Fe(IV)=O π-bond induces bond formation between the O atom of the Fe(IV)=O and the nitrogen atom of the oxidized substrate. The rate-limiting transition state barrier corresponds to the single electron transfer from the substrate into the hydroperoxo localized σ* LUMO and was calculated to be ~8 kcal/mol lower in energy than that of a non-protonated peroxo as in P. Protonation of the peroxo bridge increases the electron affinity by lowering the energy of the hydroperoxo localized σ* LUMO, lowering the reaction barrier for the single electron transfer. Correspondence between the NRVS analysis, DFT simulations, PES and RC calculations indicate that the µ-1,2-hydroperoxy structure in Figure 4C is an appropriate model for the P' intermediate[19][19] Park, K.; Li, N.; Kwak, Y.; Srnec, M.; Bell, C. B.; Liu, L. V.; Wong, S. D.; Yoda, Y.; Kitao, S.; Seto, M.; Hu, M.; Zhao, J.; Krebs, C.; Bollinger, J. M., Jr. and Solomon, E. I.: J. Am. Chem. Soc. 2017, 139, 7062-7070. (doi: 10.1021/jacs.7b02997).
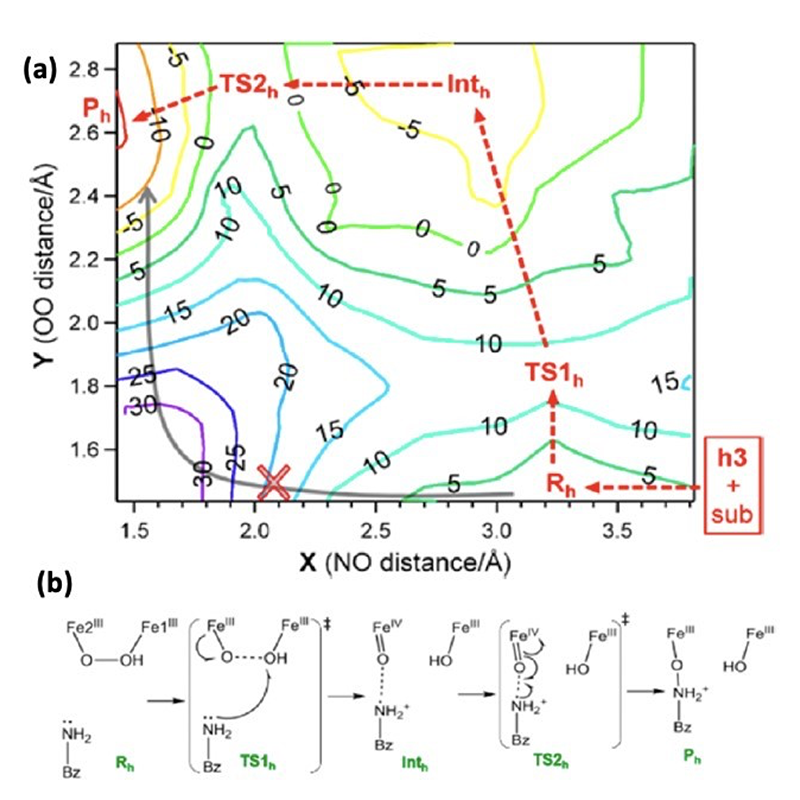
Figure 5 Calculated potential energy surface and reaction coordinate by the µ-1,2-hydroperoxo model NRVS informed model (a) potential energy surface (b) reaction coordinate.
Future Directions
NRVS has become an essential technique in evaluating oxygen intermediates in the superfamily of non-heme iron enzymes, and hence understanding the mechanisms of their diverse chemical reactivities. In the mononuclear subfamily, we are now expanding NRVS to intermediates of the intradiol dioxygenases to understand the fundamental driving forces differentiating extradiol vs. intradiol substrate cleavage. In the binuclear subclass we are using the results of our AurF study to inform a correlative study on the P' intermediate in CmlI, which shares the N-oxygenase activity of AurF while possessing an additional bridging oxo or OH- moiety. These studies are key in understanding the high reactivity of binuclear Fe(III)2 hydroperoxo species relative to the very low reactivity of the mononuclear non-heme Fe(III) hydroperoxides. We are also applying NRVS to intermediate Q in soluble methane monooxygenase (sMMO) to determine its geometric structure and its unique ability to activate the inert C-H bond of methane. These studies will continue to provide foundational insight into the exotic chemical transformations performed by the non-heme iron enzyme superfamily.
Acknowledgements
Nuclear resonance vibrational spectroscopic measurements were approved by JASRI and conducted at SPring-8 BL09XU through the long-term proposal 2017A0137 - 2018B0137. The authors thank Y. Yoda and M. Seto for assistance at the beam line. These projects were funded through the National Institutes of Health (GM040392).
References
[1] Solomon, E. I.; Brunold, T. C.; Davis, M. I.; Kemsley, J. N.; Lee, S. K.; Lehnert, N.; Neese, F.; Skulan, A. J.; Yang, Y. S. and Zhou, J.: Chem. Rev. 2000, 100, 235-350.
[2] Wong, S. D.; Srnec, M.; Matthews, M. L.; Liu, L. V.; Kwak, Y.; Park, K.; Bell, C. B., III; Alp, E. E.; Zhao, J.; Yoda, Y.; Kitao, S.; Seto, M.; Krebs, C.; Bollinger, J. M., Jr. and Solomon, E. I.: Nature 2013, 499, 320-323.
[3] Srnec, M.; Wong, S. D.; Matthews, M. L.; Krebs, C.; Bollinger, J. M., Jr. and Solomon, E. I.: J. Am. Chem. Soc. 2016, 138, 5110-5122.
[4] Srnec, M. and Solomon, E. I.: J. Am. Chem. Soc. 2017, 139, 2396-2407.
[5] Liu, L. V.; Bell, C. B., III; Wong, S. D.; Wilson, S. A.; Kwak, Y.; Chow, M. S.; Zhao, J.; Hodgson, K. O.; Hedman, B. and Solomon, E. I.: Proc. Natl. Acad. Sci. 2010, 107, 22419-22424.
[6] Sturhahn, W.: Hyperfine Interactions 2000, 125, 149-172.
[7] L. B. Gee, spectra.tools: https://www.spectra.tools/
[8] Smith, M. C.; Xiao, Y.; Wang, H.; George, S. J.; Coucouvanis, D.; Koutmos, M.; Sturhahn, W.; Alp, E. E.; Zhao, J. and Cramer, S. P.: Inorg. Chem. 2005, 44, 5562-5570.
[9] Park, K.; Tsugawa, T.; Furutachi, H.; Kwak, Y.; Liu, L. V.; Wong, S. D.; Yoda, Y.; Kobayashi, Y.; Saito, M.; Kurokuzu, M.; Seto, M.; Suzuki, M. and Solomon, E. I.: Angewandte Chemie Int-Ed 2013, 52, 1294-1298.
[10] Park, K.; Bell, C. B.; Liu, L. V.; Wang, D.; Xue, G.; Kwak, Y.; Wong, S. D.; Light, K. M.; Zhao, J.; Alp, E. E.; Yoda, Y.; Saito, M.; Kobayashi, Y.; Ohta, T.; Seto, M.; Que, L. and Solomon, E. I.: Proc. Natl. Acad. Sci. 2013, 110, 6275-6280. (doi: 10.1073/pnas.1304238110)
[11] Park, K. and Solomon, E. I.: Can. J. Chem. 2014, 92, 975-978.
[12] Liu, L. V.; Bell, C. B.; Wong, S. D.; Wilson, S. A.; Kwak, Y.; Chow, M. S.; Zhao, J.; Hodgson, K. O.; Hedman, B. and Solomon, E. I.: Proc. Natl. Acad. Sci. 2010, 107, 22419-22424.
[13] Decker, A.; Chow, M. S.; Kemsley, J. N.; Lehnert, N. and Solomon, E. I.: J. Am. Chem. Soc. 2006, 128, 4719-4733.
[14] Sutherlin, K. D.; Liu, L. V.; Lee, Y. M.; Kwak, Y.; Yoda, Y.; Saito, M.; Kurokuzu, M.; Kobayashi, Y.; Seto, M.; Que, L. Jr.; Nam, W. and Solomon, E. I.: J. Am. Chem. Soc. 2016, 138, 14294-14302.
[15] Sutherlin, K. D.; Rivard, B. S.; Böttger, L. H.; Liu, L. V; Rogers, M. S.; Srnec, M.; Park, K.; Yoda, Y.; Kitao, S.; Kobayashi, Y.; Saito, M.; Seto, M.; Hu, M.; Zhao, J.; Lipscomb, J. D. and Solomon, E. I.: J. Am. Chem. Soc. 2018, 140, 5544-5559.
[16] Groce, S. L. and Lipscomb, J. D.: Biochemistry 2005, 44, 7175-7188.
[17] Kovaleva, E. G. and Lipscomb, J. D.: Science. 2007, 316, 453-457.
[18] Sutherlin, K. D.; Wasada-Tsutsui, Y.; Mbughuni, M. M.; Rogers, M. S.; Park, K.; Liu, L. V.; Kwak, Y.; Srnec, M.; Böttger, L. H.; Frenette, M.; Yoda, Y.; Kobayashi, Y.; Kurokuzu, M.; Saito, M.; Seto, M.; Hu, M.; Zhao, J.; Alp, E. E.; Lipscomb, J. D. and Solomon, E. I.: J. Am. Chem. Soc. 2018, 140, 16495-16513.
[19] Park, K.; Li, N.; Kwak, Y.; Srnec, M.; Bell, C. B.; Liu, L. V.; Wong, S. D.; Yoda, Y.; Kitao, S.; Seto, M.; Hu, M.; Zhao, J.; Krebs, C.; Bollinger, J. M., Jr. and Solomon, E. I.: J. Am. Chem. Soc. 2017, 139, 7062-7070. (doi: 10.1021/jacs.7b02997)




