



Volume 23, No.4 Pages 335 - 342
1. 最近の研究から/FROM LATEST RESEARCH
長期利用課題報告2
P型ATPaseの結晶構造解析
Crystal Structure Analysis of P-type ATPases
東京大学 定量生命科学研究所 Institute for Quantitative Biosciences, The University of Tokyo
- Abstract
- 膜蛋白質結晶中の脂質二重膜の可視化を目指し、複数の長期利用課題を通してX線溶媒コントラスト変調法を開発してきた。4つの状態のCa2+ポンプ結晶に適用した結果、これまでは1,2分子しか見えなかったCa2+ポンプを取り囲む燐脂質すべて(~45分子)を解像できた。その結果、膜蛋白質には燐脂質の「錨」としてともに動くアミノ酸残基と膜に浮かぶための「浮き」となる残基が配置されており、両者の間に緊密な連携があること、脂質二重膜もイオン輸送メカニズムの一部として組み込まれていることが分かった。
1. はじめに
2016A期からBL41XUを利用して、長期利用課題「P型ATPaseの結晶構造解析」を、研究室のメンバー(小川治夫准教授、金井隆太助教、椛島佳樹助教)とともに遂行した。この長期利用課題はイオンポンプ蛋白質の作動機構の原子構造による完全な理解を目指すものであり、具体的には(i)15年以上にわたって追求してきた筋小胞体Ca2+ポンプ(Ca2+−ATPase、SERCA1a)の反応サイクル中間体の結晶構造解析、(ii)医学的生物学的にはより重要とも言え、そのために激しい国際競争が続いているナトリウムポンプ(Na+, K+−ATPase)の複数の状態の結晶解析、(iii)「結晶中の脂質二重膜の可視化」を行い、膜蛋白質が働く「場」である脂質二重膜とCa2+ポンプとの相互作用を解明すること、の3つのテーマからなる。それぞれ大きな進歩があったが、特に膜の可視化に関しては、歴代のBL41XUビームラインスタッフとも協力してX線コントラスト変調法を開発し、実験開始から実に15年かかったことになるが、その成果をNature誌2017年5月11日号にArticleとして発表、表紙を飾ることができた[1][1] Y. Norimatsu, K. Hasegawa, N. Shimizu and C. Toyoshima: Nature 545 (2017) 193-198.。またNews & Views[2][2] K. J. Sweadner: Nature 545 (2017) 162-164.でも単独紹介された。ここではその内容を解説したい。
2. 膜蛋白質結晶中の脂質二重膜可視化
ここ10年ほどの間に膜蛋白質そのものに関する我々の知識は飛躍的に増大したが、膜蛋白質が活動する場である脂質二重膜そのもの、或いはその構成成分である燐脂質(+コレステロール)と蛋白質の相互作用に関する知識は依然として極めて限定されている[3][3] F. M. Goñi: Biochim. Biophys. Acta 1838 (2014) 1467-1476.。これまで、膜の厚さは塩基性(正の荷電を持つ)アミノ酸残基(リジン(Lys)、アルギニン(Arg))やトリプトファン(Trp)によって影響を受け、「蛋白質-脂質ミスマッチ(予想される疎水性部分の長さが蛋白質側と脂質二重膜側で異なる)」が起こること、また、Lys・Argは膜の疎水性部分から長い側鎖を伸ばして(snorkellingと言う)燐脂質の燐酸部分と強く結合すること(図1左)[4][4] J. A. Killian and G. von Heijne: Trends Biochem. Sci. 25 (2000) 429-434.、Trpはおそらくカルボニル基と水素結合を作ること(図1右下)、また、燐脂質頭部の種類や脂肪酸の長さは安定性や活性に影響を与えることは分かっていたが、45年前に提出された「流動モザイクモデル」[5][5] S. J. Singer and G. L. Nicolson: Science 175 (1972) 720-731.からの本質的な進歩はなかったと言える。例えば、膜貫通ヘリックスの動きに燐脂質は追従するのか、或いは脂質二重膜はどこまで変形できるのか、など分かっていないことばかりであった。実験的にこのような問題にアプローチするためには結晶中の脂質二重膜を可視化することが必要である。
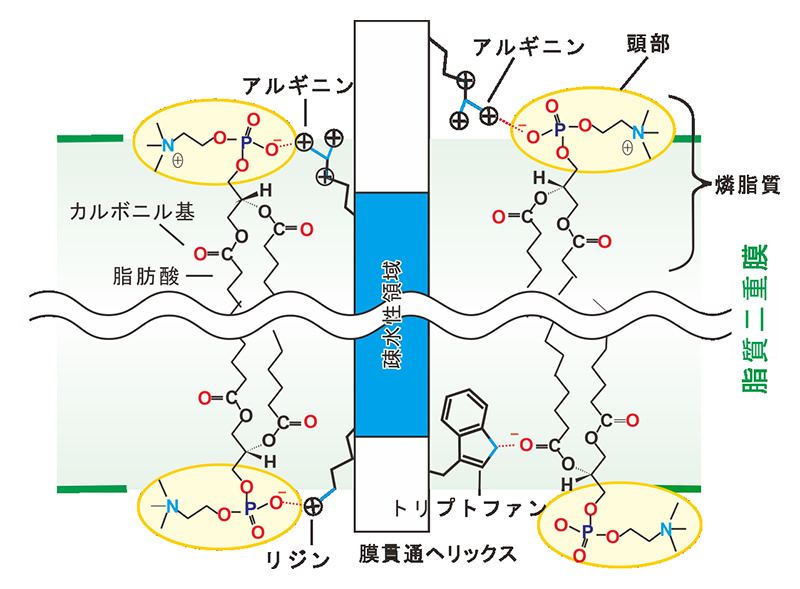
図1 アミノ酸残基と脂質二重膜を構成する燐脂質間の極性相互作用の模式図。図には代表的な燐脂質であるphosphatidylcholine(PC)を示す。燐脂質の脂肪酸部分は14−20の炭素鎖からなるが、この図では途中を省略してある。
残念ながら、通常のX線結晶解析は脂質二重膜の可視化には無力である。それには幾つかの理由がある。第一に、燐脂質の柔軟性によってその回折強度の大部分は1/10 Å−1以下の極低角部分に限定されるが(図2)、通常の結晶解析では極低角の反射強度の測定は行わないし、そもそも蛋白質の原子モデルの構築のためには不必要である。さらに、通常の解析では脂質二重膜の存在は無視し、蛋白質の外側は均一な溶媒で満たされているものと仮定して精密化を行う。これは蛋白質結晶中の脂質二重膜の現実的モデルを簡単には構築できない以上、致し方のないことではあるが、このような結晶解析はすべて誤りを含んでいることを忘れてはなるまい。

図2 構造因子に対する脂質二重膜の寄与。コントラスト変調剤iohexolの濃度が0%(黒線)と45%(紫線)の場合。蛋白質の電子密度はiohexol濃度45%の溶媒とほぼ一致するので脂質二重膜の寄与は最も顕著になり、Bragg spacing 10 Åではiohexolの効果が明らかである。なお、Bragg spacing 15 Å(0.07 Å−1)を越える低角領域では脂質二重膜の寄与は構造因子全体の50%を上回ることに注意されたい。
それでは、(0,0,1)反射のような最低角からのすべての反射強度を測定すれば十分かというとそうではない。このような低角領域では脂質二重膜に由来する反射強度は蛋白質に由来する強度より大きくなるため(図2)、反射点の位相は蛋白質に由来する位相とは大きく異なるからである。実際、すべての反射点を用い、蛋白質原子モデル由来の位相で電子密度図を書いてみると、膜は見事に消えている(図3(b))。部分的に低角を入れたときに見える電子密度(図3(c))は、堂々と論文になったりしているが[6][6] Y. Sonntag, M. Musgaard, C. Olesen, B. Schiøtt, J. V. Møller, P. Nissen and L. Thøgersen: Nature Comm. 2 (2011) 304.、雑音に過ぎない可能性が高い。
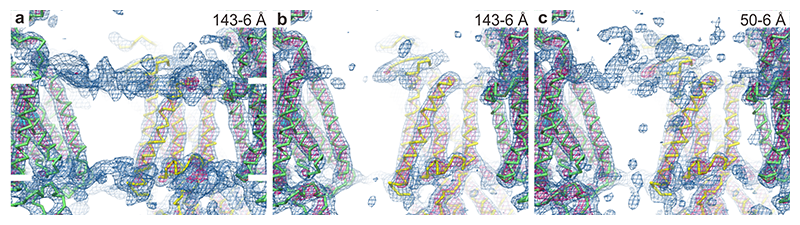
図3 Ca2+ポンプ結晶(E1~P・ADP・2Ca2+状態)の膜貫通領域の電子密度図に対する最低分解能領域の構造因子の影響。電子密度図に寄与した反射の分解能を示す。電子密度図は0.7 σで、図の右上に示した領域の反射を用いて描き、脂質二重膜の膜面に並行に見ている。(a) 溶媒コントラスト変調由来の位相を用いた場合。(b),(c) 原子モデルに由来する位相を用いた場合。(a)の白い横棒は燐脂質の燐酸機部分のおよその位置を示す。
実際、通常の結晶解析では、蛋白質の膜貫通領域と直接接触している第一層の燐脂質すら解像されず、蛋白質のセグメント間(膜貫通ヘリックス間の隙間とか、隣接する蛋白質分子との隙間とか)に挟まれて固定されている場合にのみ解像される。すなわち、アクアポリン0の二次元結晶[7][7] T. Gonen et al.: Nature 438 (2005) 633-638.のような例外的な場合を除き、第一層の燐脂質のせいぜい2,3分子が解像されるに過ぎない[8][8] N. D. Drachmann, C. Olesen, J. V. Møller, Z. Guo, P. Nissen and M. Bublitz: FEBS J. 281 (2014) 4249-4262.。このため、低角の位相を正しく決定できれば個々の燐脂質分子を解像できるはずだという確信は、筆者にしてもまったくなかった。
3. 脂質二重膜の可視化によって理解されたこと
脂質二重膜を可視化しようと考えたのは、不完全な結晶解析に不満だったためだけではない。私たちはCa2+ポンプSERCA1aを対象に、その反応過程のほぼ全体をカバーする10状態の結晶構造を決定し、イオン能動輸送機構の原子構造に基づく理解を推進してきた[9][9] C. Toyoshima: Arch. Biochem. Biophys. 476 (2008) 3-11.(図4)。その結果、膜貫通ヘリックスは膜に垂直な方向にも10 Å以上動くこと、脂質二重膜はポンプのメカニズムの一部として組み込まれているらしいことは理解された[10][10] C. Toyoshima: Biochim. Biophys. Acta 1793 (2009) 941-946.。しかし、例えば、そのような膜貫通ヘリックスの大きな動きに対し燐脂質は追随して一緒に運動するのか、そもそも二重膜はどれほど凸凹しているのか、10本ある膜貫通ヘリックスのうちM7−M10はほとんど構造変化を起こさないから反応サイクル中、ずっと同じ向きで膜に挿入されると考えてよいのか、といった基本的疑問には答えるすべがなかった。従って、イオンポンプの原子構造に基づく機能の理解のためには、少なくとも、第一層の燐脂質分子すべてを解像し相互作用の詳細を知る必要がある、つまり、当たり前のことなのだが、膜蛋白質を理解するためには脂質二重膜をも知る必要があり、可視化のための実験的手法を確立する必要があると確信するに至った。
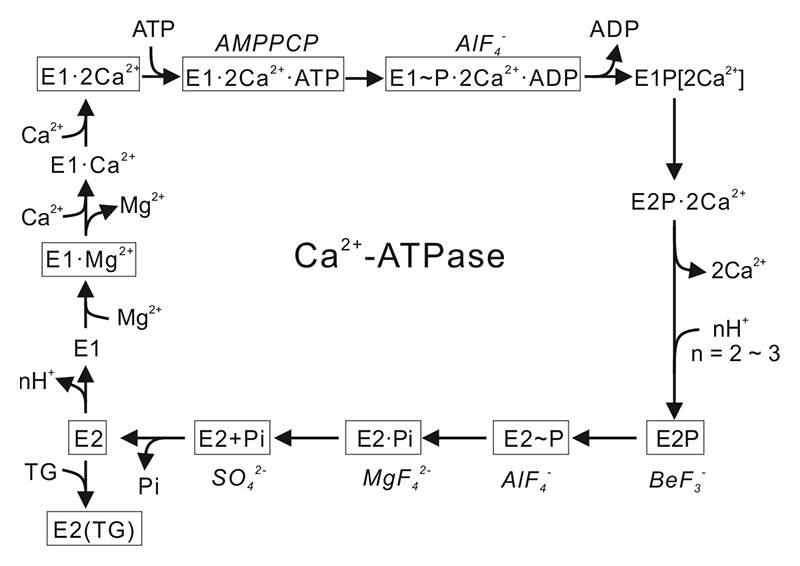
図4 Ca2+ポンプ(SERCA1a)の反応ダイアグラム。四角で囲った状態は構造決定済み。四角の外にはその状態を安定化するために用いた基質アナログを示す。
そこで、X線溶媒コントラスト変調を利用することを考え、方法論的開発とデータ収集を行ってきた。その結果、Ca2+ポンプの4つの状態(Ca2+結合直後のE1・2Ca2+、ATPから蛋白質への燐酸転移反応の遷移状態であり細胞質側ゲートは閉位置にあるE1~P・ADP・2Ca2+、膜内に結合していたCa2+を小胞体内腔側に放出した直後であり蛋白質に結合した燐酸の加水分解反応の遷移状態であるE2~P、Ca2+を放出し終わった後の安定状態E2)(図4参照)に関し、脂質二重膜を可視化することができた[1][1] Y. Norimatsu, K. Hasegawa, N. Shimizu and C. Toyoshima: Nature 545 (2017) 193-198.。これまでの結晶解析では蛋白質の隙間に挟まれた脂質1,2分子しか解像できなかったが[8][8] N. D. Drachmann, C. Olesen, J. V. Møller, Z. Guo, P. Nissen and M. Bublitz: FEBS J. 281 (2014) 4249-4262.、Ca2+ポンプを取り巻く個々の燐脂質をすべて(蛋白質1分子あたり約45分子)解像することに成功し、燐脂質頭部の原子モデルを置くことができた。さらに、低角の位相はコントラスト変調から、高角の位相は原子モデルからとなるように位相を結合することで、3.2 Å分解能での通常の結晶学的精密化にも成功し(図5(a))、分子動力学シミュレーションにより得られた原子モデルを確認することもできた(図6(b))。この結果、ポンプ蛋白質と燐脂質の相互作用の詳細が明らかになり、これまで燐脂質を同じように固定すると考えられてきたLys/ArgとTrp/Tyrは明確に異なった役割を持ち協働していることなど、まったく新しい知見が得られた。すなわち、脂質二重膜は膜蛋白質を浮かべている単なる海のようなものではなく、機能発現に能動的に係わっていること、そのため、燐脂質をも含む系全体の原子モデルからメカニズムを考える必要があることが明確になった。今や、蛋白質を構成するアミノ酸残基に通し番号がつけられているように、Ca2+ポンプに関しては、燐脂質にも通し番号をつけて議論できるようになったのである(図5)。
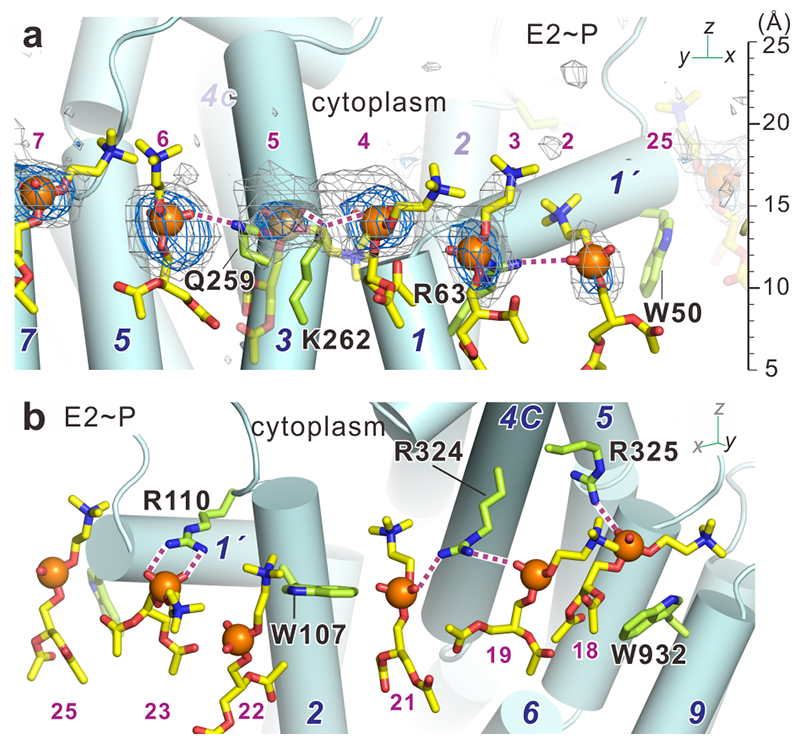
図5 Ca2+−ポンプの膜貫通領域を取り囲む燐脂質(E2~P状態)。(a) X線溶媒コントラスト変調による3.2 Å分解能での2|Fo|-|Fc|電子密度図(青色のネット1.0 σ、灰色のネット0.7 σ:低角は溶媒コントラスト、高角は原子モデルによる位相を結合したもの)とそれに基づく燐脂質の原子モデル(phosphatidylcholineの頭部からカルボニル基までを棒で、オレンジ色の小球で燐原子を示す)。水色の円筒とイタリックの数字は膜貫通ヘリックス(M1−M10)を示し、紫色の小さい数字は本研究によって付与された燐脂質のIDである。アミノ酸残基の標記はKがリジン、Qがグルタミン、Rがアルギニン、Wがトリプトファンに対応する。紫色の破線は水素結合の候補を、(a)の欄外のものさしは脂質二重膜の中心からの距離を示す。(b)は(a)とほぼ同じ領域を反対側から見たもの。(a)では正の荷電を持つアミノ酸残基(R63、K262はその例)は膜内の疎水性部分から側鎖を伸ばして(snorkelling)負の荷電を持つ燐脂質の燐酸基と強く結合し、燐脂質の「錨」となる。この場合、膜貫通ヘリックスの運動とともに燐脂質は上下運動する。(b)のように細胞質側から燐酸基と結合する場合は、「錨」として(コンフォメーション変化のスイッチとして)燐脂質を利用しており(R110、R324、R325はその例)、反応サイクル中の状態の遷移によるヘリックスの運動に際し相手となる燐脂質を変える。トリプトファンは溶媒と脂質の界面に存在し、燐脂質頭部からカルボニルの位置まで広く分布する(図1のようにカルボニル基と水素結合を作るとは限らない)。
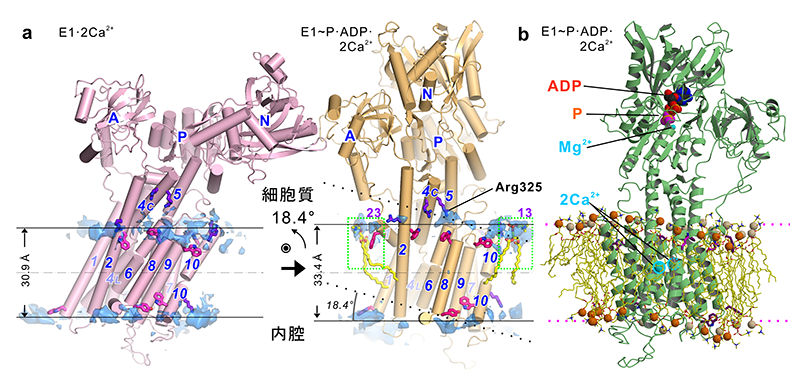
図6 反応サイクル中のCa2+ポンプ分子全体の配向の変化。(a) ATP結合と燐酸の転移による脂質二重膜に対する配向の変化。水色の塊はコントラスト変調で得られた脂質二重膜の電子密度(燐脂質頭部に対応)を示す。4.5 Å分解能で計算し、0.44 e−/Å3で描いた。水平線(実線)は得られた燐脂質の燐原子の、点線はM7−M10ヘリックスの膜に対する配向は変わらないとしたときに予想される燐原子の平均位置を示す。E1・2Ca2+ → E1~P・ADP・2Ca2+(結合したCa2+を膜内に閉じ込める過程)では分子全体が18.4°傾斜する。両親媒性ヘリックスM1'周辺(PL23)とM10付近の燐脂質分子(PL13)の原子モデルをスティック(緑四角)で示す。赤い棒はトリプトファン、青の棒は塩基性残基を、斜体数字は膜貫通ヘリックスの番号(M1−M10)を示す。(b) Ca2+ポンプと脂質二重膜の原子モデル(E1~P・ADP・2Ca2+状態)。燐脂質頭部は結晶構造解析によって、脂肪酸部分は100 nsの分子動力学シミュレーションによって得られたもの。オレンジ色の球は燐原子を示す。(a)とは見ている方向が反対。
以下に主な結果をまとめる。
・膜は決して平らではなく(図5)、厚さ(燐脂質頭部の燐酸基間の距離)も(30.9~33.4 Å:図6)、蛋白質を取り囲む燐脂質の数(44~48)も反応サイクル中で変化する。それに伴って最適の位置に戻そうとする力が働くはずで、その力は構造変化の駆動力として使われている可能性がある。
・膜貫通ヘリックスの運動に伴い、膜内から側鎖を伸ばして(snorkellingして)いるLys/Argと結合している燐脂質(図1、5)は一緒に動く。すなわち、ヘリックスの膜面に対し垂直な方向の運動に追従し局所的な歪みを生み出す。それは、ヘリックスを元の位置に戻す力として働くだろう。
・一方、膜外(細胞質側)から燐脂質と結合するArgは相手となる燐脂質を変え、その燐脂質をコンフォメーション変化の際の「錨」として使っている(図1右上、図5のR324)。Arg324の変異体、特にGlu変異体ではCa2+ポンプのATP加水分解反応は著しく阻害されるが、その理由は、他の残基との相互作用がないためこれまで理解できなかった[11][11] K. Yamasaki, T. Daiho, S. Danko and H. Suzuki: J. Biol. Chem. 279 (2004) 2202-2210.。
・膜に対する蛋白質の向きを決めるのは主としてTrpである(図5)。Trpは溶媒と脂質の界面に存在し[12][12] W. M. Yau, W. C. Wimley, K. Gawrisch and S. H. White: Biochemistry 37 (1998) 14713-14718.、「浮き」の役割を持つ。これまでに想定されていたように燐脂質のカルボニル基と水素結合を作る(図1右下)わけではなく、燐酸基からカルボニル基までの広い範囲をカバーする(四重極相互作用によるらしい[13][13] S. Chakravarty, A. R. Ung, B. Moore, J. Shore and M. Alshamrani: Biochemistry 57 (2018) 1852-1867.)ので、膜面に沿ったヘリックスの大きな(> 20 Å)動きにも適している(例えば、E2 → E1・2Ca2+遷移におけるM1'やM2ヘリックスの運動)。
・膜貫通ヘリックスの運動に伴って、ヘリックスの疎水性部分が溶媒に露出しないように、また親水性部分が脂質二重膜の疎水性部分に露出しないように、蛋白質側は複雑な構造変化を起こす。特に注目すべきは、分子全体の傾きが20°近く変化することである(図6)。このときには、Trp4残基からなるベルトが形成され、いつも膜面と平行になっていた。Trpが脂質二重膜の両方の層にあるのはM10付近に限られ、そこが分子全体の傾斜の際の軸となっていると考えられる。膜に垂直な方向へ大きく動くのは分子のほぼ反対側にあるM2であるから、これは、分子全体の小さな傾きの変化で膜貫通ヘリックスの大きな運動を許すためのメカニズムと考えられる。
4. 溶媒コントラスト変調法
以下では脂質二重膜の可視化のための方法論を少し議論しておきたい。ここでの具体的課題は、1/10 Å−1より低角の反射に対し正しい実験的位相を与えることである。この目的のためには2つの方策が考えられ、1つは標準的な(i)多重同型置換法(multiple isomorphous replacement: MIR)であり、もう1つは(ii)溶媒コントラスト変調法(solvent contrast modulation)である。(i)の多重同型置換法では、重原子を含む溶液に蛋白質結晶を浸漬し、異なる位置に結合した重原子による反射強度の変化から位相を決定する。X線結晶解析における位相決定のための標準的方法であるが、極低角での位相決定には幾つかの問題点がある。元来、分子量10万を越える蛋白質による回折振幅を分子量100程度の重原子が変化できる割合はごく小さい。特に、本課題の場合、脂質二重膜からの寄与は蛋白質からの寄与の2倍程度あり(図2)、重原子による寄与はさらに小さくなる。より深刻かもしれない問題は、重原子が非特異的に或いは小さい占有率で多数の場所に結合することであり、それがどの程度反射強度に影響を与えるかは見積もる手段がない。実際、重原子の添加によって、その重原子が特定の場所に結合するかどうかによらず、溶媒の電子密度は変化し、各反射点における散乱強度は溶媒の電子密度の二次函数として変化する(図7(b))。また、通常の結晶化条件下では二次函数の極小からは大きくずれているため、散乱強度は溶媒の電子密度の変化に敏感である(コントラスト効果)。
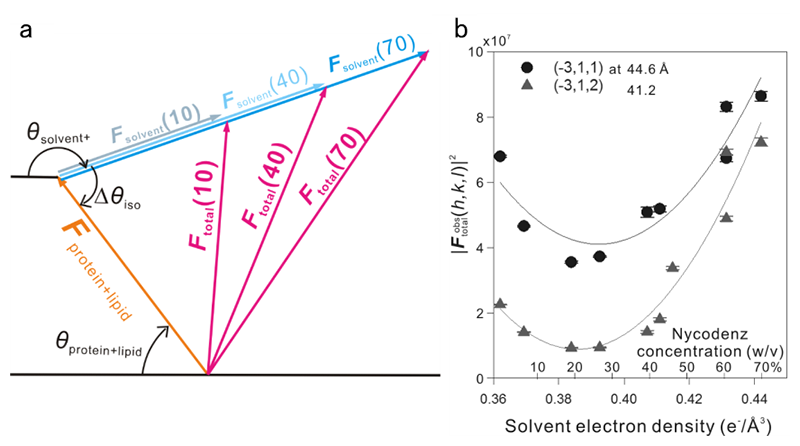
図7 (a) 実測される回折振幅と、蛋白質+脂質二重膜の構造因子、溶媒の構造因子との関係。コントラスト変調剤の濃度ξ = 10, 40, 70%の場合について示す。各回折点の回折強度は(b)に示すように、溶媒の電子密度<ρsolvent>(ξ)の二次函数として変化する。ここでθsolventは溶媒の構造因子Fsolventの位相部分、など。(b) acentric反射(-3,1,l)における構造因子の2乗|F(-3,1,l)|2を溶媒電子密度の函数としてプロットしたもの。線は最少二乗法による理論曲線。コントラスト変調剤の濃度が低いところでは回折振幅は溶媒の電子密度変化に敏感であることに注意。
溶媒コントラスト変調法は、蛋白質の概形の決定手段として開発されたが、中性子回折により膜蛋白質結晶中の界面活性剤ミセルを可視化するための標準的な方法となった[14][14] E. Pebay-Peyroula, R. M. Garavito, J. P. Rosenbusch, M. Zulauf and P. A. Timmins: Structure 3 (1995) 1051-1059.。溶媒の散乱能(X線であれば電子密度)を変えれば、対象とするものの見え方は大きく変わる。もし蛋白質を取り囲む溶媒の密度を蛋白質の密度と一致させれば、蛋白質と溶媒は区別がつかなくなり、密度が異なる他の部分(界面活性剤のミセルや脂質二重膜など)だけが見えるため、モデリング可能になる。しかし、精密化の方法はなく、情報量はごく少なかった。一方、このような変化は、回折強度の変化として反映されるだろうから(図7)、その変化分を差し引くことができれば、固定部分(つまり蛋白質+脂質二重膜)の密度情報が得られると期待できる。だが、溶媒密度変化による回折強度変化から出てくる位相(⊿θiso)は、必要としている試料全体、或いは固定部分の位相と直接的には結びついていない(図7(a))。従って、コントラスト変調から出てくる位相は、答えが正しいかどうかの検証には使えるにしても、直接的には使い難い。そのため、初期位相(つまりは脂質二重膜のモデル)がどうしても必要となるが、それを用いて初期電子密度図を計算できれば、蛋白質部分と溶媒部分のflatteningを使って、位相改良が可能なはずである。
ここで極めて有効であったのが、溶媒置換率(Pexと表記する)という概念を導入したことである[1][1] Y. Norimatsu, K. Hasegawa, N. Shimizu and C. Toyoshima: Nature 545 (2017) 193-198.。試料中のすべての点の電子密度は溶媒の電子密度に依存せず一定であるか、溶媒電子密度の一次函数として直線的に変化すべきであると考えられる(図8)。その傾きをPexとし、蛋白質や脂質二重膜の内部ではPexは0であり、溶媒部分は1、境界領域では0と1の間の値をとるとする。まず、蛋白質部分の電子密度は原子モデルから計算される値にし、溶媒部分の平均電子密度は溶媒の密度を実測して得られた値に設定する。脂質二重膜に関しては膜に垂直な方向のみに電子密度が変化する一次元単純モデルからスタートする[1][1] Y. Norimatsu, K. Hasegawa, N. Shimizu and C. Toyoshima: Nature 545 (2017) 193-198.。Pexのマップを計算し、蛋白質部分は0に、溶媒部分は1に値を制限するとともに、平滑化することによって雑音を減らすことができる。これによって、データセット間の整合をとることができるようになり、データの冗長性を位相改良にうまく結びつけることができた。

図8 溶媒置換率Pex。溶媒コントラスト変調では加えるコントラスト変調剤の濃度を変えて溶媒の電子密度を変化させる。結晶全体の電子密度分布から各濃度での溶媒と蛋白質の電子密度を差し引くことで脂質二重膜の電子密度図が得られる。その際、各点における電子密度は、蛋白質や膜部分では変化せず(Pex = 0)、溶媒部分や境界領域では各点の溶媒置換率Pexに応じて溶媒密度の一次函数として変化すべきであることを利用する。0−80%のiohexolで変化できる電子密度(0.1 e−/Å3)を図中に示した。
コントラスト変調剤としては幾つかの可能性が考えられる。脂質二重膜の可視化を目指す以上、燐脂質頭部の燐酸基と結合する物質は避けたい。従って、正の荷電を持ったイオン性物質は最初の試みから除外すべきであろう。Fourmeらは電子顕微鏡法で実績のあるaurothioglucoseを提案していた[15][15] W. Shepard, R. Kahn, M. Ramin and R. Fourme: Acta Crystallogr. D 56 (2000)1288-1303.。我々も試したが、この化合物は照射損傷が激しく溶解度も高くない上に、結晶格子を破壊することが判明した。分子量の小さい物質は分解能の点からはよさそうなのだが、分子間の隙間に深く入りこむ結果、結晶格子を破壊する傾向が強いようである。実際、非常に有用であることが判明したのは、iohexolである(図9:Histodenzとしてシグマから売られている)[16][16] D. Rickwood, T. Ford and J. Graham: Anal. Biochem. 123 (1982) 23-31.。X線造影剤として開発されたものであり、密度勾配遠心などにも使われる。水に対する溶解度は極めて高く80%(w/v)を越えるが可変できる電子密度は0.1 e−/Å3に過ぎず、実効的な分解能は5 Å程度にとどまると考えられる[1][1] Y. Norimatsu, K. Hasegawa, N. Shimizu and C. Toyoshima: Nature 545 (2017) 193-198.。
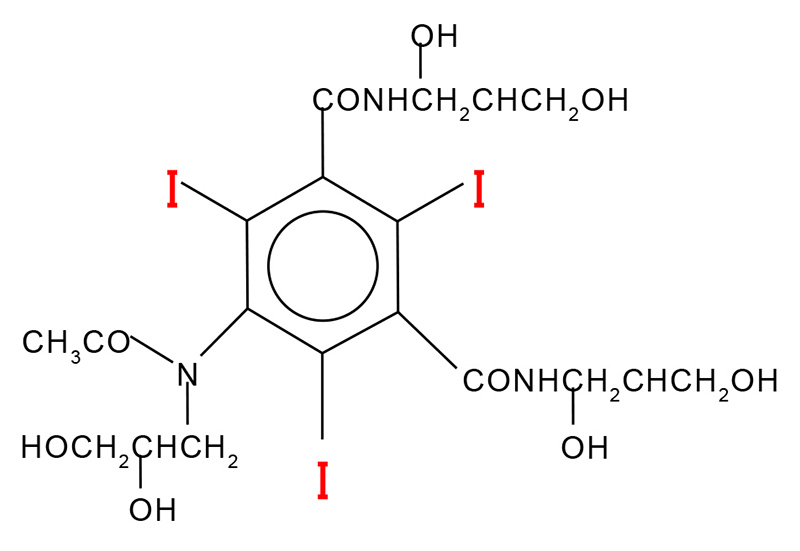
図9 iohexolの化学構造[16][16] D. Rickwood, T. Ford and J. Graham: Anal. Biochem. 123 (1982) 23-31.。この物質は水に80%(w/v)可溶であり、溶媒電子密度を約0.10 e−/Å3上昇させることができる。
このようにX線コントラスト変調ではコントラストの可変範囲が狭く、図8のように、x軸に関しては極めて狭い範囲のデータから直線の傾きとy切片を求めることになっている。従って、高精度の回折強度データの測定が要求される。幸運であったことは、Ca2+ポンプの結晶化に必要であったpolyethylene glycolの濃度は10%程度と低く、多くの場合、iohexol濃度70%(w/v)を越えることができたため、溶媒コントラスト変調の可変範囲は比較的広かったことである。測定精度の問題もあるが、結晶化条件が、蛋白質の電子密度とほぼ一致し、コントラストを逆転できる40−50% iohexolを許容できることは最低限必要である。一方、iohexolを用いた場合、その大きさから、5 Åより高分解能の回折点の強度はiohexolの濃度に依存しないと考えられる。従って、原子モデルを用いた絶対スケーリングが可能のはずである。実際、高分解能で構造決定したCa2+ポンプ蛋白質の原子モデルを用い、初期的な温度因子の補正や同型性の評価も可能であった。これは、X線を用いることの非常に大きな利点である。そのため、データ収集にあたっては、分解能3.2 Å程度までのデータを精度良く集める必要がある。
一方で、溶媒領域を決定するマスクの問題は深刻である。現状の精密化プログラムでは原子モデルの外側を一定の電子密度で埋めることしかできない。従って、蛋白質+脂質二重膜領域の中は原子モデルで埋まらない限り、真空である。且つ乱れた構造(「一様な密度」と言ってもよいが)を原子モデルで表現するのは極めて困難である。そのために、部分的な脂質二重膜の原子モデルでは、通常の精密化プログラムを用いた場合、極低角まで含めるとR因子はかえって悪くなる。そのため、電子密度図(図5(a))の計算では、極低角はコントラスト変調による位相を、高角は原子モデルによる位相を用いるようにphase combineした構造因子を使う必要がある。
5. データ収集に関して
最初期(2002年頃)の予備的実験はBL40B2を用いて行ったが、有用なデータが得られるようになったのは、BL41XUで、当時のビームライン担当者であった清水伸隆博士、酒井久伸博士の協力を得てHeパスを設置し、カメラ長600 mm、イメージングプレートX線検出器R-Axis Vを用いることによってすべての反射点の回折データを収集できるようになってからである(2004年、図10)。原子モデルを用いたスケーリングのために必要な3~4 Å分解能の反射点とともに、検出器を飽和させることなく極低角の反射強度を測定しようとすると、検出器のdynamic rangeは16 bitでは使い物にならず、20 bitでも露光を1/10以下にして再露光する必要があった。そのために、CCDが検出器として使えるようになってもR-Axis Vを使う必要があり、異様に時間のかかる実験であった(結晶系がC2であったため、180フレーム × 2分 = 6時間)。その後、二次元フォトンカウンティング検出器PILATUS3 6Mが導入され、測定時間そのものは大幅に(10分以下に)短縮されたが、やはり部分的には2~3条件での測定が必要なフレームがあり、長期利用課題がなければ実行不可能な研究であった。

図10 Ca2+ポンプ結晶(E2(TG)状態)のX線回折パターン。60%(w/v)のiohexolを含む緩衝液に浸漬したもの。BL41XUでR-Axis Vイメージングプレート検出器を用い3.0 Å分解能まで記録した。カメラ長600 mm、波長1 Å。この結晶の対称性はP41212であり、格子長は、a = b = 71.56、c = 586.14 Åである。右下には四角で囲んだ領域の拡大を示す(但し、露光は落としてある)。
理論的にはすべての反射点の回折強度は溶媒電子密度の二次函数として変化するので[15][15] W. Shepard, R. Kahn, M. Ramin and R. Fourme: Acta Crystallogr. D 56 (2000)1288-1303.、1つの状態の構造決定には最低3つ(実際には10点程度)の溶媒密度での測定が必要である(図7(b))。結晶はすべて微量透析法で得られたものであり、再現性も非常に良好であった。1つの成分だけの変化は容易であり、且つ、厳密な制御が可能であったことも大きかった。結晶を拾うときの乾燥の問題も低温室(10ºC)で行うことで解決できた。但し、高濃度のiohexol存在下で結晶を拾うことは、溶媒が相当に粘稠になり、且つコントラストマッチングのために結晶が見えなくなるなど、ある程度の熟練を要するのですべて筆者が行った。
6. おわりに
本研究で用いたX線溶媒コントラスト変調法は、大きな揺らぎを持った構造を可視化するために有用な方法であるが万能ではない。初期位相を必要とするし、高濃度のpolyethylene glycolを沈殿剤として結晶化している場合には、溶媒コントラスト変調の余地はない。さらに、コントラスト変調剤の大きさによる分解能の問題があり、溶媒の電子密度の可変範囲が狭いことによる精度の問題もある。そのような問題のない、H2O/D2Oによるコントラスト変調が可能な中性子回折への発展が期待されるが、ビーム強度の問題はまだ深刻であり、巨大結晶が必要である。そこで、多重同型置換法のデータ収集を工夫して低角の位相情報を得る手段を確立することも考えている。いずれにせよ、低角の位相決定の問題を克服し、脂質二重膜からプロトンまでを含む真の構造生物学を目指している。そのための方法論を今後の長期利用課題の中で追求したい。
謝辞
本研究はSPring-8の長期利用課題(2009B0025、2013A0049、2016A0133)の一部と一般課題2012B1486としてなされたものである。コントラスト変調の解析ソフトウェアは研究室の乗松良行博士とともに開発したものである。回折データ収集にあたってはSPring-8 BL41XUの歴代の担当者、特に、清水伸隆博士、酒井久伸博士、長谷川和也博士、奥村英夫博士の絶大なご支援をいただいた。ここに記して御礼申し上げたい。また、結晶を作製してくれた杖田淳子さん、平田絢美さん、岩澤志穂さん、データ収集を手伝ってくれた研究室のメンバーに感謝したい。
参考文献
[1] Y. Norimatsu, K. Hasegawa, N. Shimizu and C. Toyoshima: Nature 545 (2017) 193-198.
[2] K. J. Sweadner: Nature 545 (2017) 162-164.
[3] F. M. Goñi: Biochim. Biophys. Acta 1838 (2014) 1467-1476.
[4] J. A. Killian and G. von Heijne: Trends Biochem. Sci. 25 (2000) 429-434.
[5] S. J. Singer and G. L. Nicolson: Science 175 (1972) 720-731.
[6] Y. Sonntag, M. Musgaard, C. Olesen, B. Schiøtt, J. V. Møller, P. Nissen and L. Thøgersen: Nature Comm. 2 (2011) 304.
[7] T. Gonen et al.: Nature 438 (2005) 633-638.
[8] N. D. Drachmann, C. Olesen, J. V. Møller, Z. Guo, P. Nissen and M. Bublitz: FEBS J. 281 (2014) 4249-4262.
[9] C. Toyoshima: Arch. Biochem. Biophys. 476 (2008) 3-11.
[10] C. Toyoshima: Biochim. Biophys. Acta 1793 (2009) 941-946.
[11] K. Yamasaki, T. Daiho, S. Danko and H. Suzuki: J. Biol. Chem. 279 (2004) 2202-2210.
[12] W. M. Yau, W. C. Wimley, K. Gawrisch and S. H. White: Biochemistry 37 (1998) 14713-14718.
[13] S. Chakravarty, A. R. Ung, B. Moore, J. Shore and M. Alshamrani: Biochemistry 57 (2018) 1852-1867.
[14] E. Pebay-Peyroula, R. M. Garavito, J. P. Rosenbusch, M. Zulauf and P. A. Timmins: Structure 3 (1995) 1051-1059.
[15] W. Shepard, R. Kahn, M. Ramin and R. Fourme: Acta Crystallogr. D 56 (2000)1288-1303.
[16] D. Rickwood, T. Ford and J. Graham: Anal. Biochem. 123 (1982) 23-31.
東京大学 定量生命科学研究所
〒113-0032 東京都文京区弥生1-1-1
TEL : 03-5841-8492
e-mail : ct@iam.u-tokyo.ac.jp




