



Volume 19, No.2 Pages 96 - 101
1. 最近の研究から/FROM LATEST RESEARCH
リチウムイオン電池正極材料の非平衡な相変化挙動を世界で初めて観察
Direct Observation of Nonequilibrium Phase Transition of Lithium Ion Battery Cathode
京都大学大学院 人間・環境学研究科 Graduate School of Human and Environmental Studies, Kyoto University
- Abstract
- 結晶構造・電子構造変化をリアルタイムで観測可能な「時間分解X線吸収分光法」「時間分解X線回折法」をリチウムイオン電池動作環境で適用させ、高速充放電時における電極材料の相変化挙動解明を試みた。SPring-8の高輝度放射光は、充放電反応中の構造情報を連続的に取得することを可能にした。高速充放電可能な正極材料であるLiFePO4は、充放電反応中には熱力学的に安定な二つの相の間で反応することが知られている。高速充放電中の結晶構造を「時間分解X線回折法」により解析した結果、安定な二相に加え、中間の格子定数を有する中間相LixFePO4相が生成していることを初めて発見した。LixFePO4相の出現量は反応速度に比例し、定常状態では消滅することから、準安定な相であることが判明した。準安定なLixFePO4相は、高速反応時に生成し、二相の歪みを緩和することで、LiFePO4の高速充放電特性を発現していると考えられる。
1. はじめに
エネルギーを蓄えるデバイスとして蓄電池やキャパシタが挙げられるが、中でも高エネルギー密度を有するリチウムイオン電池が高い注目を集めている。リチウムイオン電池は1991年に実用化されて以来、エネルギー密度やエネルギー変換効率の高さから、高性能蓄電池としてモバイル機器の電源などに広く用いられ、社会の発展に貢献してきた。近年、環境・エネルギー問題の観点から、電気自動車用電源、電力消費の負荷平準化などの更なる需要が生まれ、性能向上を目指し熾烈な研究開発が行われている。社会からの需要は極めて高いものの、リチウムイオン電池の反応機構は未だにブラックボックスな点を残しており、その解明による発展の余地が大きく残っているといえる[1][1] J. M. Tarascon and M. Armand, Nature, 414 (2001) 359-367.。
リチウムイオン電池は充電時に正極からLi+が脱離し、電解質中を拡散し、負極に挿入されることで酸化還元反応のエネルギーが蓄えらえる。放電時には逆の反応が起こり、電子の流れから電気を取り出すことが出来る。正極にLiCoO2、負極に黒鉛を用いるときの充電反応は次式で表される。
| 6C + xLi+ + xe–→LixC6 | (1) |
| LiCoO2→Li1–xCoO2 + xLi+ + xe– | (2) |
リチウムイオン電池の充放電反応では、複数の時間スケール、空間スケールにまたがってそれぞれの反応が平行して進行している。この反応に伴う階層構造をFig. 1に示す。まず、充放電に伴い電極/電解質界面におけるLi+の相間イオン移動の反応が起こる。相間イオン移動過程は非常に短い時間に小さい空間領域で起こるため、Li+移動反応のメカニズムは未解明な部分が多く、その解明により二次電池の性能向上が期待できると考えられる。
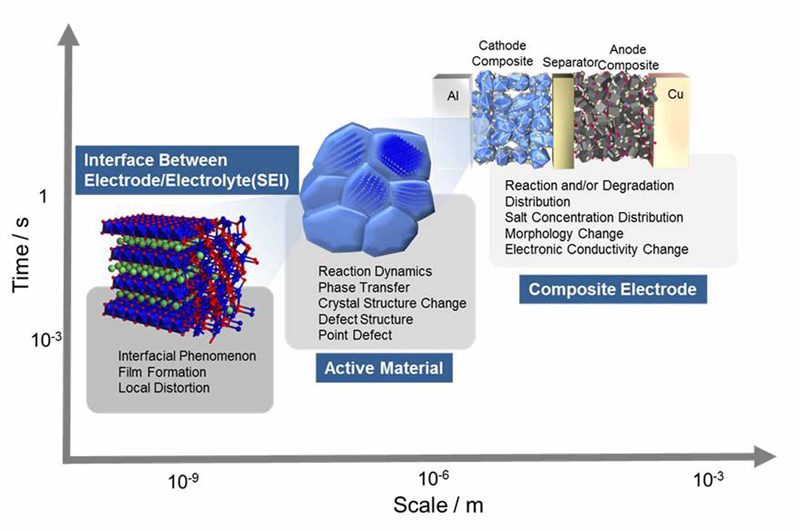
Fig. 1 リチウムイオン二次電池の反応における時間・空間スケール模式図
界面を通過したLi+は電極活物質内を拡散する。結晶内のLi量変化は電極材料の構造変化を引き起こす。インターカレーション電極では、LiCoO2のように相転移せずに材料の格子定数が連続的に変化する固溶反応系電極と、LiFePO4のようにLi-richな相とLi-poorな相に相分離する二相反応系電極に大別される。実際の電池作動下においてはこのような変化が連続して起こるため非平衡状態の活物質の構造変化を把握する事は重要であるが、これまでの反応解析は平衡状態をベースにしたものがほとんどであり、十分な知見は得られていない。
活物質の粒は実際には導電剤、結着剤と混合して合材電極として用いられることから、更に大きな合材電極のスケールで考えることも必要である。例えば電極の面方向や深さ方向に反応が起こりやすい場所の分布が生じる可能性が考えられる。同じ粒ばかりが反応に使われてしまうと、劣化が進行し、電池の寿命が縮まってしまう。
リチウムイオン電池の性能向上のためには、このようなマルチスケールの現象を統合的に理解することが重要である。SPring-8で使用可能な高輝度放射光は、上記の異なるスケールに対応した現象解明に有効なツールである。本研究では、上記で述べた階層構造のうち、活物質相変化の非平衡挙動解明に注目した。
相変化挙動を解析するための活物質として、LiFePO4を選択した。LiFePO4は比較的安価なFeを主成分としている点から、低コストであること、PO4骨格による結晶構造の安定性が高く、サイクル特性、安全性に優れている利点を有している[2][2] A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 144 (1997) 1188-1194.。本材料は報告された当初、低い電子伝導性のため、出力特性が不十分である問題があったが、微粒子化[3][3] A. Yamada, S. C. Chung and K. Hinokuma, J. Electrochem. Soc., 148 (2001) A224-A229.、炭素被覆[4][4] S. Y. Chung, J. T. Bloking and Y. M. Chiang, Nat. Mater., 1 (2002) 123-128.のプロセスにより、既存のLiCoO2系を上回る出力特性が実現可能となっている。LiFePO4は充放電反応において、Li-rich相(LFP相)とLi-poor相(FP相)に相分離し、二相の割合のみが変化する事で全体のLi量が変化することが報告されている[5][5] A. Yamada, H. Koizumi, S. I. Nishimura, N. Sonoyama, R. Kanno, M. Yonemura, T. Nakamura and Y. Kobayashi, Nat. Mater., 5 (2006) 357-360.。二相共存反応におけるLFP相/FP相の相転移機構は、様々なモデルが考案されている。LiFePO4を正極材料として最初に報告したPadhiらは、LiFePO4結晶子の表面で核生成が起こり、Liの挿入脱離が等方的に進行しながら相境界が粒内部に進行する、shrinking coreモデルを提案した[2][2] A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 144 (1997) 1188-1194.。しかし、Li+拡散が一次元方向に異方的に進行することが報告されると[6][6] D. Morgan, A. Van der Ven and G. Ceder, Electrochem. Solid State Lett., 7 (2004) A30-A32.、反応が等方的に進行するshrinking coreモデルは疑わしくなった。また球の表面で核生成が起こる場合、二相の境界面積が大きくなり、歪みエネルギーが大きくなってしまうことからもshrinking coreモデルは考え難い。そこでshrinking coreモデルに変わる、異方性のある反応機構モデルが提案された。ChenらはLiを脱離させた鱗片状のLiFePO4をb軸方向からTEM観察し、二相の相境界からb軸方向にLiが拡散しながら相境界がa軸方向に移動するという異方的な新たなモデルを提案した[7][7] G. Y. Chen, X. Y. Song and T. J. Richardson, Electrochem. Solid-State Lett., 9 (2006) A295-A298.。Delmasらはこのモデルを拡張し、核生成速度よりも核成長速度の方が圧倒的に速いと考え、一度核が生成すると相境界が速やかに移動して結晶全体が相転移するという "Domino-cascade" モデルを提案した[8][8] C. Delmas, M. Maccario, L. Croguennec, F. Le Cras and F. Weill, Nat. Mater., 7 (2008) 665-671.。このモデルは有望な反応機構モデルとして高い支持を集めているが、引き続き新たなモデルが提案され続けている[9-11][9] N. Meethong, Y. H. Kao, W. C. Carter and Y. M. Chiang, Chem. Mater., 22 (2010) 1088-1097.
[10] R. Malik, F. Zhou and G. Ceder, Nat. Mater., 10 (2011) 587-590.
[11] P. Bai, D. A. Cogswell and M. Z. Bazant, Nano Lett., 11 (2011) 4890-4896.。
このようにLiFePO4の相転移挙動は明確でなく、今もなお新たなモデルが考案され続けているのが現状である。相転移挙動が解明されていない理由は、今まで挙げられたモデルのほとんどが平衡状態の測定から、もしくは計算から、非平衡状態を推測しているためである。実際の充放電反応でどのように相が変化するかを知ることは新規材料の設計指針を得るためにも重要であり、問題の解決が待たれている。本研究では放射光を用いた高速時分割測定の適用により二相共存系正極LiFePO4における未知の相転移挙動を直接観測することで、高速充放電特性を有する正極材料の特徴を明らかにした[12-14][12] Y. Orikasa, T. Maeda, Y. Koyama, H. Murayama, K. Fukuda, H. Tanida, H. Arai, E. Matsubara, Y. Uchimoto and Z. Ogumi, Chem. Mater., 25 (2013) 1032-1039.
[13] Y. Orikasa, T. Maeda, Y. Koyama, H. Murayama, K. Fukuda, H. Tanida, H. Arai, E. Matsubara, Y. Uchimoto and Z. Ogumi, J. Am. Chem. Soc., 135 (2013) 5497-5500.
[14] Y. Orikasa, T. Maeda, Y. Koyama, T. Minato, H. Murayama, K. Fukuda, H. Tanida, H. Arai, E. Matsubara, Y. Uchimoto and Z. Ogumi, J. Electrochem. Soc., 160 (2013) A3061-A3065.。本研究では今まで計算や定常状態の測定からの推測でしか議論されてこなかった非平衡状態の相挙動について解明し、二相共存系電極の一般的な特性解明に繋がる見識を得られる点で重要である。
2. 実験方法
水熱法により1000 nmの粒径を有するLiFePO4を作製した。活物質、導電材、結着剤を重量比75:15:10で混合したものを集電体のAl箔上に塗布して電極を作製し、負極、参照極にLi金属、電解液に1 mol LiPF6(ethylene carbonate:ethyl methyl carbonate = 3:7 vol)を用い、放射光測定可能な三極式のラミネートセルを作製した。放射光時間分解測定として、X線吸収分光測定(XAS)、X線回折測定(XRD)を行った。それぞれの測定におけるセットアップ模式図をFig. 2に示す。時分割XASはBL01B1、BL28XUにて行った。測定は透過法にてFe K-edgeを測定した。ラミネートセルと測定系を同期させ、充放電反応とスペクトル取得を同時に行った。充放電レートは6分での充放電に対応する10 Cレート(全容量を1時間で充電もしくは放電する電気量を1 Cレートと言い、その何倍かをn Cレートで表記)に設定した。1スペクトルあたりの計測時間は7秒であり、15秒に1スペクトルのXAS測定を連続的に行った。時分割XRDはBL28XU、BL46XUで行った。回折計の中心にセルをセットし、二次元検出器PILATUSにて回折線を測定した。波長0.9995 Å、露光時間0.5秒で、XRD測定を行った。露光時間以外はシャッターでX線を遮断することでセルダメージの影響を排除した。LiFePO4は二相領域の外側に単相反応の領域が存在する[5][5] A. Yamada, H. Koizumi, S. I. Nishimura, N. Sonoyama, R. Kanno, M. Yonemura, T. Nakamura and Y. Kobayashi, Nat. Mater., 5 (2006) 357-360.。この影響を取り除くため、本実験においては、あらかじめ放電状態から3.35 Vまで充電反応を進行させた上で、測定を開始した。充放電反応の上限電位は4.3 V、下限電位は2.0 Vとし、5サイクルの充放電サイクル中にXAS、XRD測定を行った。
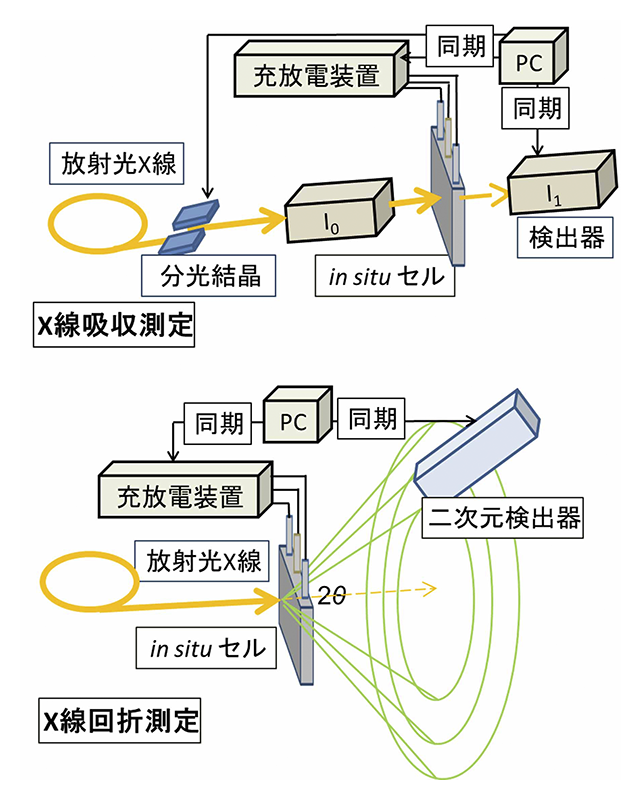
Fig. 2 時間分解X線吸収分光法およびX線回折測定のセットアップ模式図
3. 結果と考察
Fe K-edgeのXASスペクトルはFeの平均価数を反映する。LiFePO4系に時分割XASを適用する事で、LiFePO4(Fe2+)からFePO4(Fe3+)への変化を観測する事ができる[15][15] A. Deb, U. Bergmann, S. P. Cramer and E. J. Cairns, Electrochim. Acta, 50 (2005) 5200-5207.。粒径1000 nmのLiFePO4の10 C充放電反応中XASスペクトルをFig. 3に示す。充電反応に伴い吸収端が高エネルギー側へシフト、放電時に低エネルギー側へのシフトが観測された。これは充放電にともなうリチウムの挿入脱離により、
| LiFePO4⇔FePO4 + Li+ + e– | (3) |
の反応が進行していることに由来する。各スペクトルはLFP相とFP相のスペクトルの足し合わせで表すことができる。レート10 Cで充放電反応を5サイクル繰り返し、15分間緩和させた間の各スペクトルにおける二相の割合をFig. 4に示す。XASから算出した値(プロット)と電流量からの求めた値(点線)は非常に良い一致を示した。つまり、電気化学的に制御される電子のやりとりは即座に活物質中の価数状態に反映されることを意味している。また、電流量とFe中の価数変化量が対応していることから、セル中において副反応が起こっておらず、正常な充放電反応が進行していることを示すものである。
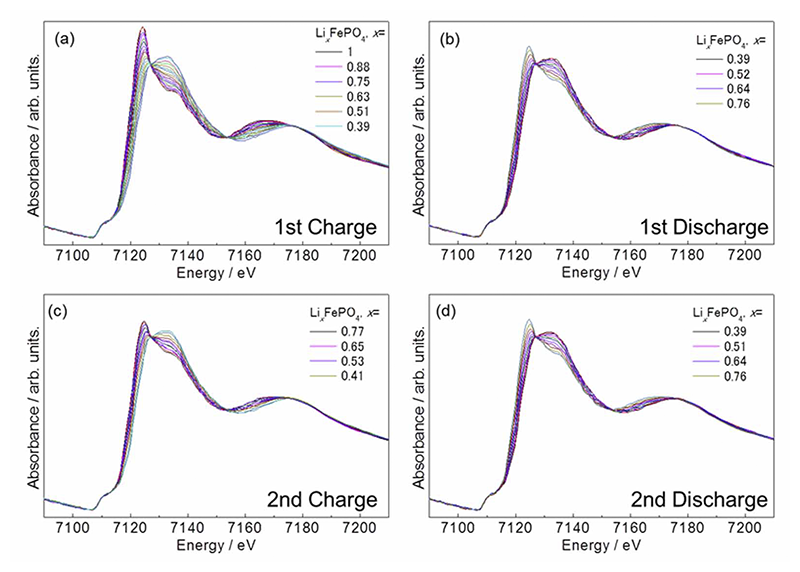
Fig. 3 10 Cレートにて充放電サイクル中のFe K-edge XASスペクトル[13][13] Y. Orikasa, T. Maeda, Y. Koyama, H. Murayama, K. Fukuda, H. Tanida, H. Arai, E. Matsubara, Y. Uchimoto and Z. Ogumi, J. Am. Chem. Soc., 135 (2013) 5497-5500.
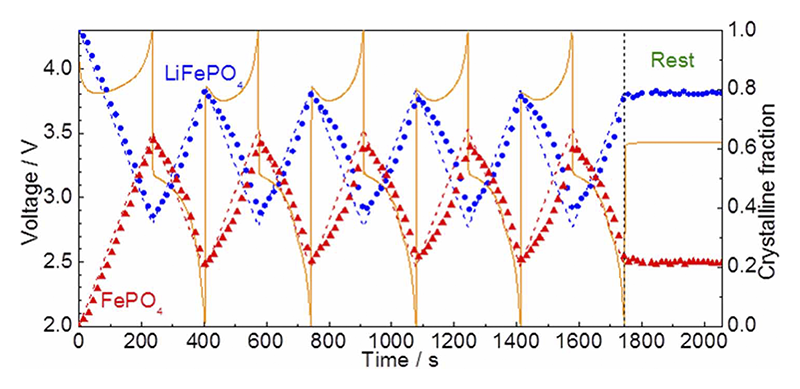
Fig. 4 10 Cレートにて充放電サイクル中のX線吸収スペクトルから算出したLiFePO4、FePO4相の割合(プロット)と電気量から推定される相変化量(点線)。橙線は充放電プロファイル[13][13] Y. Orikasa, T. Maeda, Y. Koyama, H. Murayama, K. Fukuda, H. Tanida, H. Arai, E. Matsubara, Y. Uchimoto and Z. Ogumi, J. Am. Chem. Soc., 135 (2013) 5497-5500.
一方で結晶相の変化は熱力学的に想定される二相反応とは全く異なる挙動を示した。レート10 Cで充放電反応を5サイクル繰り返し、15分間緩和させた間の時間分解XRDプロファイルをFig. 5に示す。19.15˚付近のピークがLFP相の211面、020面に、19.5˚付近のピークがFP相の211面に、19.85˚付近のピークがFP相の020面に相当する。充電反応進行に伴いLFP相のピークが減少し、FP相のピークが増加することから、二相共存反応が進行していることが分かる。これに加えて、19.35˚付近にLFP相、FP相由来ではない新相のピークが出現した。新相ピークは最初の充電過程では出現しないが放電過程において成長し、続く充電過程で消滅する。
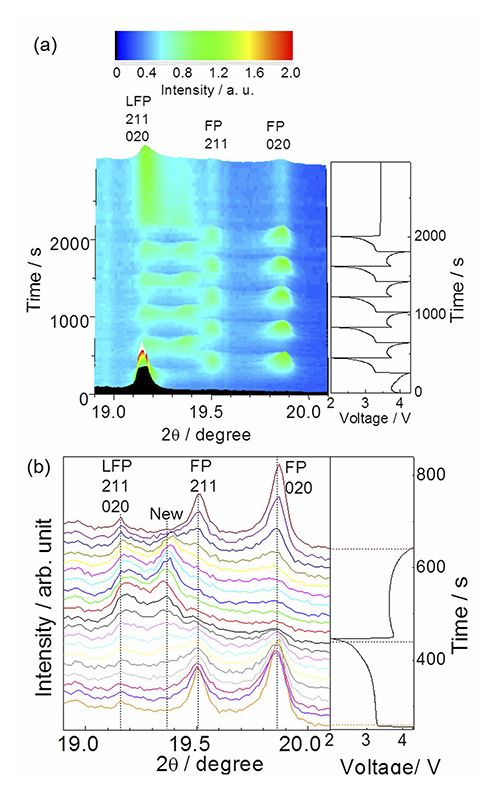
Fig. 5 レート10 Cにおいて充放電サイクル中の時間分解XRDパターンおよび充放電曲線[13][13] Y. Orikasa, T. Maeda, Y. Koyama, H. Murayama, K. Fukuda, H. Tanida, H. Arai, E. Matsubara, Y. Uchimoto and Z. Ogumi, J. Am. Chem. Soc., 135 (2013) 5497-5500.
1本の回折線からでは新たに出現したものが何であるかの議論は困難である。そこで、10 C充放電中、他の角度範囲においても時分割XRD測定を行った。200、301回折線について測定した、反応開始から2nd充電終了までの時分割XRDをFig. 6に示す。200、301回折線においても放電時に同様の新相生成が確認できた。020、200、301面の新相のピークがLiFePO4と同じ斜方晶であると仮定して格子定数を算出したところ、a = 10.21 Å、b = 5.945 Å、c = 4.750 Åとなり、新相はLFP相、FP相の間の格子定数をとることが判明した。以後このLixFePO4相をLxFP相と定義する。本研究で発見されたLxFP相の格子定数は、高温XRDの結果として報告されている固溶相LixFePO4(x = 0.6~0.75)の値に極めて近くなった[16-17][16] G. Y. Chen, X. Y. Song and T. J. Richardson, J. Electrochem. Soc., 154 (2007) A627-A632.
[17] C. Delacourt, P. Poizot, J. M. Tarascon and C. Masquelier, Nat. Mater., 4 (2005) 254-260.。したがって、高温において安定に見られる固溶相LixFePO4(x = 0.6~0.75)が室温における電気化学反応中の非平衡状態においても生成すると考えられる。

Fig. 6 LiFePO4の10 C充放電サイクル中における200、301のXRDパターン[13][13] Y. Orikasa, T. Maeda, Y. Koyama, H. Murayama, K. Fukuda, H. Tanida, H. Arai, E. Matsubara, Y. Uchimoto and Z. Ogumi, J. Am. Chem. Soc., 135 (2013) 5497-5500.
このLxFP相の生成は速度依存性が存在する。Fig. 7は異なる充放電レートにて同様の実験を行った際、最もLxFP相が生成する初期充放電直後のXRDパターンをプロットしたものである。LxFPのピークは充電レートが増加するにつれて明確に確認できるようになる。同組成の状態で24時間以上緩和した場合のXRDパターンではLxFPのピークは全く見られなくなった。以上から、LxFP相は充放電速度が速い場合に優先的に成長し、平衡状態では観測することができない準安定相であることが判明した。
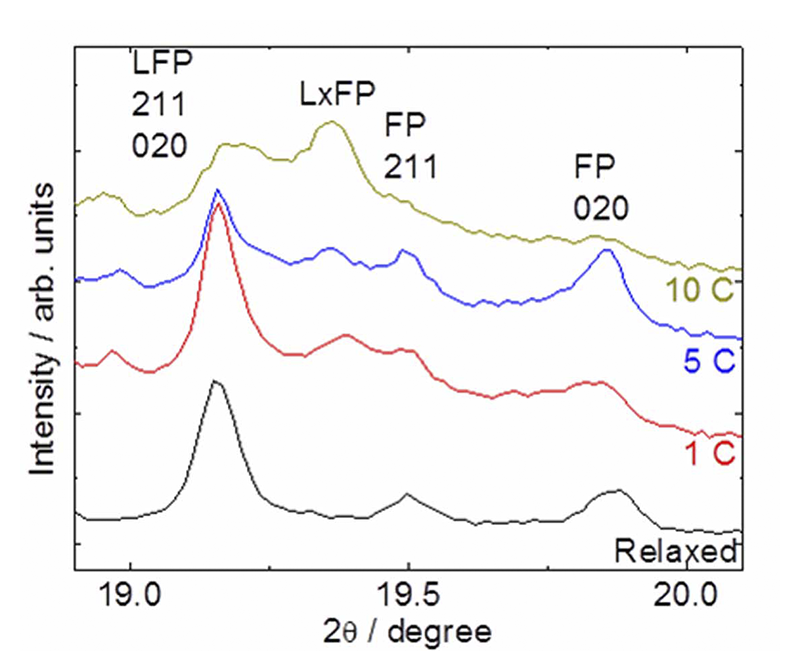
Fig. 7 LxFP相生成の充放電速度依存性[13][13] Y. Orikasa, T. Maeda, Y. Koyama, H. Murayama, K. Fukuda, H. Tanida, H. Arai, E. Matsubara, Y. Uchimoto and Z. Ogumi, J. Am. Chem. Soc., 135 (2013) 5497-5500.
時間分解測定の結果をもとにLiFePO4の相転移モデルをFig. 8に整理する。FP相からLFP相への相転移について、反応電位がFP/LFPの平衡電位を下回るとき、FPからLFPへの転移が起こる(Fig. 8(b)Aの領域)。電流が小さい場合はこの準静的な過程、つまり熱力学的に推定される二相共存反応により相転移が進行する。LiFePO4系の反応の律速過程は核生成であることが示唆されている[8][8] C. Delmas, M. Maccario, L. Croguennec, F. Le Cras and F. Weill, Nat. Mater., 7 (2008) 665-671.。FP/LFP二相の格子定数ギャップはb軸で3.6%、c軸で1.8%と大きいため、核生成に伴う界面エネルギーが大きくなる。したがってFPからLFPに直接転移する反応では大きな電流は得ることが難しい。大きな出力つまり、大電流での反応を進めるためには準安定相LxFP相の存在が有意となる。LxFP相は熱力学的に準安定であるため、FP/LxFPの反応電位はFP/LFPの反応電位よりも低くなる(Fig. 8(b)Bの領域)が、充放電過程では電気化学的にその過電圧分を補うことが可能である。FPとLxFPの格子ミスマッチはb軸で2.5%、c軸で0.66%とLFPの場合より小さいため、LxFP相の核生成が起こりやすくなる。このようにして、核生成律速であるLiFePO4において、パスAで示すような中間相LxFPを経由した核生成が起こることで反応が滞りなく進行することが可能となる。本現象は、リチウムイオン二次電池作動条件下でのみ観測されるユニークな現象であり、LiFePO4が優れた高速充放電特性を示す理由の一つであると推定される。
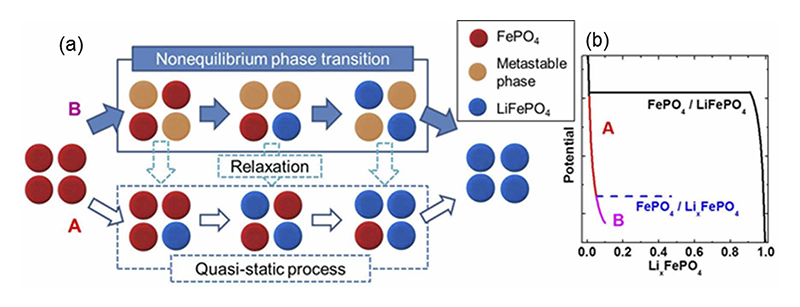
Fig. 8 (a)高速充放電中の相変化メカニズムのモデル図 (b)相変化とエネルギーの関係[13][13] Y. Orikasa, T. Maeda, Y. Koyama, H. Murayama, K. Fukuda, H. Tanida, H. Arai, E. Matsubara, Y. Uchimoto and Z. Ogumi, J. Am. Chem. Soc., 135 (2013) 5497-5500.
4. まとめと今後の展望
本研究ではLiFePO4の高速充放電中において二相の中間の格子定数を有する新相LixFePO4相が生成することを初めて発見した。LixFePO4相の出現量は反応速度に比例し、二相の歪みを緩和するLixFePO4相が二相の中間に生成する事で反応が滞りなく進行する事が、LiFePO4が高速充放電特性を有する要因であると考えられる。時間分解測定によるLiFePO4の相転移挙動の研究は、今まで計算や定常状態の測定からの推測でしか議論されてこなかった非平衡状態の相挙動について直接的な観察を行い、未知の相転移現象を明らかにしたものであり、新規二相反応系電極の設計指針の構築に端緒を与える結果を得られた点で重要である。
SPring-8には蓄電池解析にターゲットを絞り、産官学共同で研究開発を進めるためのビームラインBL28XUが運用されている。これまで定常状態解析、解体分析を主としていた蓄電池解析は、実際の充放電条件下で直接的に状態観測する方向へ向かっており、蓄電池の理論性能を最大限発揮するための解析手法が、SPring-8では世界にさきがけて整備されている。本研究で確立された解析スキームは、産業界の蓄電池解析へ適用されており、蓄電池産業の更なる発展に貢献するものと期待される。
謝辞
本研究はNEDO「革新型蓄電池先端科学基礎研究事業(RISING)」の一環として行われました。共同研究者である京都大学の小久見善八教授、内本喜晴教授、松原英一郎教授、荒井創教授、小山幸典准教授、谷田肇准教授、福田勝利准教授、村山美乃助教にこの場を借りて深く御礼申し上げます。放射光実験は、SPring-8 BL01B1、BL28XU、BL46XU(課題番号2012A7601、2011B1034、2011B1908、2011A1014、2010B1896にて実施)にて実施されたものです。
参考文献
[1] J. M. Tarascon and M. Armand, Nature, 414 (2001) 359-367.
[2] A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 144 (1997) 1188-1194.
[3] A. Yamada, S. C. Chung and K. Hinokuma, J. Electrochem. Soc., 148 (2001) A224-A229.
[4] S. Y. Chung, J. T. Bloking and Y. M. Chiang, Nat. Mater., 1 (2002) 123-128.
[5] A. Yamada, H. Koizumi, S. I. Nishimura, N. Sonoyama, R. Kanno, M. Yonemura, T. Nakamura and Y. Kobayashi, Nat. Mater., 5 (2006) 357-360.
[6] D. Morgan, A. Van der Ven and G. Ceder, Electrochem. Solid State Lett., 7 (2004) A30-A32.
[7] G. Y. Chen, X. Y. Song and T. J. Richardson, Electrochem. Solid-State Lett., 9 (2006) A295-A298.
[8] C. Delmas, M. Maccario, L. Croguennec, F. Le Cras and F. Weill, Nat. Mater., 7 (2008) 665-671.
[9] N. Meethong, Y. H. Kao, W. C. Carter and Y. M. Chiang, Chem. Mater., 22 (2010) 1088-1097.
[10] R. Malik, F. Zhou and G. Ceder, Nat. Mater., 10 (2011) 587-590.
[11] P. Bai, D. A. Cogswell and M. Z. Bazant, Nano Lett., 11 (2011) 4890-4896.
[12] Y. Orikasa, T. Maeda, Y. Koyama, H. Murayama, K. Fukuda, H. Tanida, H. Arai, E. Matsubara, Y. Uchimoto and Z. Ogumi, Chem. Mater., 25 (2013) 1032-1039.
[13] Y. Orikasa, T. Maeda, Y. Koyama, H. Murayama, K. Fukuda, H. Tanida, H. Arai, E. Matsubara, Y. Uchimoto and Z. Ogumi, J. Am. Chem. Soc., 135 (2013) 5497-5500.
[14] Y. Orikasa, T. Maeda, Y. Koyama, T. Minato, H. Murayama, K. Fukuda, H. Tanida, H. Arai, E. Matsubara, Y. Uchimoto and Z. Ogumi, J. Electrochem. Soc., 160 (2013) A3061-A3065.
[15] A. Deb, U. Bergmann, S. P. Cramer and E. J. Cairns, Electrochim. Acta, 50 (2005) 5200-5207.
[16] G. Y. Chen, X. Y. Song and T. J. Richardson, J. Electrochem. Soc., 154 (2007) A627-A632.
[17] C. Delacourt, P. Poizot, J. M. Tarascon and C. Masquelier, Nat. Mater., 4 (2005) 254-260.
京都大学大学院 人間・環境学研究科
〒606-8501 京都市左京区吉田二本松町
TEL : 075-753-6850
e-mail : orikasa.yuuki.2a@kyoto-u.ac.jp




