





Volume1 No.1
Section B : Industrial Application Report
微結晶核延伸法により高強度化したバイオベースポリマー繊維の超小角X線散乱測定による高次構造解析
High Ordered Structural Analysis of High-Strength Bio-Based Fibers Processed by a New Drawing Method with Small Crystal Nuclei Using Ultra-Small Angle X-ray Scattering
a東京大学大学院農学生命科学研究科, b信州大学繊維学部
aGraduate School of Agricultural and Life Sciences,The University of Tokyo,bFaculty of Textile Science and Technology, Shinshu University
- Abstract
生分解性を有する微生物産生ポリエステルの1つであるポリ[(R)-3-ヒドロキシブチレート-co-4-ヒドロキシブチレート]に新規延伸法の微結晶核延伸法を適用することで、繊維内部に無数のマイクロサイズのポアが存在する特殊な繊維を作製できた。微結晶核形成時間の異なる未延伸および延伸繊維の超小角X線散乱測定を行った結果、未延伸繊維では等方的な散乱が得られ、微結晶核形成時間の増加に伴って散乱強度が増加する傾向が確認できた。一方、延伸繊維では赤道線上にストリーク状の散乱が観察され、微結晶核形成時間の増加に伴ってストリークの幅と散乱強度が増加する傾向が確認できた。このストリーク散乱は繊維軸に平行な細長いポアに起因するものと考察できた。微結晶核形成時間を制御して非晶質繊維中の微小なポアの量を変化させた後に延伸を行うことによって、ポア形状を制御できることが見出された。
キーワード:バイオベースポリマー、微生物産生ポリエステル、繊維、ポーラス、超小角X線散乱
背景と研究目的:
二酸化炭素による温暖化、石油枯渇、プラスチック廃棄物等の環境問題のため、生分解性を有する環境調和型のバイオベースポリマーに関する研究開発が盛んに行われている。その1つである微生物産生ポリエステルのポリヒドロキシブチレート(PHB)は汎用樹脂のポリプロピレン(PP)と同程度の融点(180℃)やガラス転移点(Tg)(4℃)を持つため、PPの代替材料として注目され半世紀近く研究が行われてきた。しかし、PPに比べて融点と熱分解点(220℃)が近いために成形加工がしにくい。また、結晶化が遅くTgが室温以下であるために2次結晶化による経時劣化が起こる等の弱点があるため、硬くて脆い材料であり実用化は困難であった。このような欠点を解決するために、第2成分モノマーを共重合させて物性を改質する研究や延伸加工技術の開発が行われている。
最近、我々は新たな延伸技術として微結晶核延伸法を開発した[1]。これは結晶化の遅いPHBのための手法で、溶融押出した樹脂をTg付近で急冷し、保冷することで急激な結晶化を抑制し、微小な結晶核を形成させ、その結晶核を起点として分子鎖を高配向させる延伸法である。この手法によりPHB共重合体の引張強度1.3 GPaを有する高強度繊維作製に世界で初めて成功した。小角散乱測定(BL45XU, 波長: 0.1 nm, カメラ長: 2.5 m)により、数 nm ~ 100 nm程度の範囲の構造情報が得ることができ、微結晶核形成させずに延伸した繊維ではラメラ結晶と非晶の長周期に由来する散乱が観測された(図1(a))。一方、24時間微結晶核形成させた後に延伸した繊維では長周期由来の散乱強度が弱まり、赤道方向にストリーク状の散乱が観測された(図1(b))。

図1 PHB共重合体繊維の小角散乱像延伸倍率:10倍, 微結晶核形成時間:0時間 (a)と24時間 (b) [1]
これは内部構造の違いに由来すると考えられX線トモグラフィー測定(BL47XU, 波長: 0.15 nm)が行われ、延伸繊維内部にマイクロサイズの無数の細長いポアが形成されることが確認された(図2)[2]。しかし、どの様にポアが形成されるのか、その機構の詳細は明らかになっていない。

図2 微結晶核延伸法で作製したPHB共重合体繊維のX線トモグラフィー断面像 (a)と3次元像 (b) [2]
通常の小角測定では100 nm以上は測定範囲外であり、マイクロポアの形状やサイズを定量することは難しく、X線トモグラフィー測定の分解能では500 nm以下のポアサイズの定量は困難である。そこで、数十 nm ~ 1 μmの範囲の構造情報を得ることが可能な超小角散乱測定(カメラ長: 40 m)を行い、微結晶核形成条件の違いによる散乱への影響を詳細に調べることでポア形成機構を解明することを目的とした。
実験:
PHB共重合体の1つである、ゴム状で生体適合性に優れたポリ[(R)-3-ヒドロキシブチレート-co-4-ヒドロキシブチレート]を溶融押出し、Tg近傍である氷水中で巻き取ることで非晶質繊維を作製した。氷水中で一定時間静置し、微結晶核を形成させた後に室温で手回し延伸機を用いて延伸し、熱処理を行った。氷水中での微結晶核形成時間を4 ~ 72時間と変化させた、未延伸繊維および3倍延伸繊維を作製した。
作製した繊維に対し、BL19B2での超小角X線散乱測定を行った。第2ハッチに測定試料をセットし、第3ハッチに2次元検出器(PILATUS2M)をセットした。カメラ長は40 mとし、入射光は18 keV (波長(λ): 0.069 nm)を用いた。ビームサイズは幅が300 μm、高さが150 μm であり、6秒 ~ 5分間露光した。散乱ベクトルqの大きさにして0.005 ~ 0.1 nm-1 (実空間(d)では1000 ~ 60 nm)の範囲について測定を行った。ここで2θを散乱角とするとq = (4π / λ) sinθ と定義できる。カメラ長の較正はニワトリの腱由来のコラーゲン繊維と、粒径1 μmのシリカ粒子を分散させたフィルムを用いて行った。透過光の強度をイオンチャンバーで測定し、各試料のX 線吸収率を算出して散乱強度の規格化に用いた。測定に用いた繊維は、散乱強度を高めるために複数本を束ねてフォルダーに貼り付け、繊維のエッジ部分からの寄生散乱を低減するために繊維の密度(1.3 g/cm3)と同程度の密度を有し、X線吸収の少ないグリセロールを塗布した。
散乱像からのデータ処理にはFit2D[3]を用いた。1次元化した強度データを各試料のX線吸収率により規格化し、同条件で測定した空気散乱像から1次元化した強度データをバックグラウンドデータとして減算することで各試料の強度データを得た。
結果および考察:
微結晶核形成4,24,72時間の未延伸繊維および3倍延伸繊維の超小角X線散乱像を図3に示した。

図3 超小角X線散乱像 (a)〜(c)未延伸繊維、(d)〜(f)3倍延伸繊維、 (a), (d)微結晶核形成時間4時間、(b), (e)24時間、(c), (f)72時間
いずれも垂直方向が繊維軸方向である。未延伸繊維(a) ~ (c)では等方的な散漫散乱が得られたのに対し、3倍延伸繊維(d) ~ (f)では赤道線上にストリーク状の散漫散乱が観察された。ストリーク散乱が赤道方向、つまり繊維軸に垂直方向に伸びていることから、繊維軸方向に細長いポアが形成されていることが考察できた。(d) ~ (f)を比較すると微結晶核形成時間が4時間から72時間まで増加するのにしたがって、赤道方向へのストリーク散乱の幅が増大していることが確認できた。つまり、微結晶核形成時間の増加に伴ってポアの形状がより細長く変化したことが考察できた。
未延伸および3倍延伸繊維の赤道線上の散乱強度I(q)の散乱ベクトルqに対する強度プロファイルを図4に示した。いずれも小角側で単調減少関数を示し、回折ピークなどは観察されなかった。ポアサイズに由来する回折ピークが得られなかった原因は、ポア形状の不均一さゆえにブロードになり観測できなかったのではないかと考えているが、詳細は不明である。
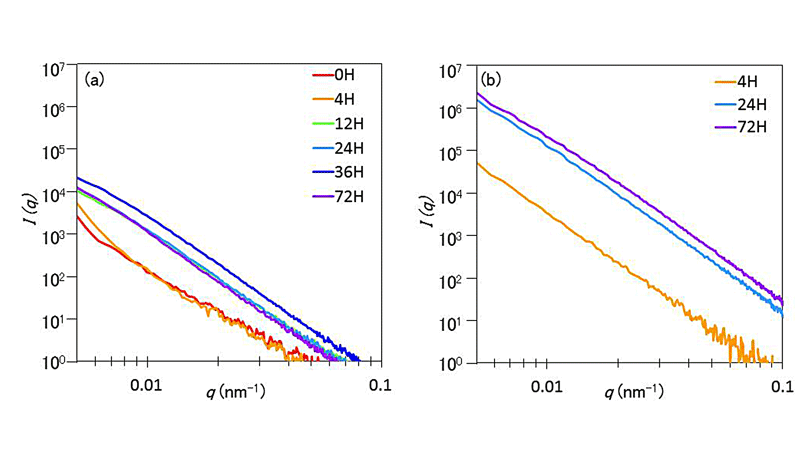
図4 超小角X線測定による赤道方向の散乱プロファイル(a)未延伸繊維、(b)3倍延伸繊維
未延伸繊維(a)では微結晶核形成時間36時間までは時間の増加に伴って、散乱強度が増大する傾向が確認できたが、72時間でわずかに散乱強度が減少した。減少した理由については不明であり、再現性も含めて検討する必要がある。未延伸繊維で微結晶核形成時間の増加に伴う散乱強度の増大から、未延伸繊維中にも微小なポアが存在し、微結晶核形成時間の増加に伴ってポアが増加したことが示唆された。図1(a) ~ (c)に示したように未延伸繊維の散乱像は等方的であることから形成されたポアは等方的に広がっていると考えられる。わずかではあるが、未延伸繊維ではq > 0.01 nm-1の領域における傾きが微結晶核形成時間の増加に伴って緩やかになっている。これがポアサイズまたはポア形状とどのような相関があるのかについても再現性も含めて検討する必要がある。
延伸繊維(b)では微結晶核形成時間4時間から24時間に増加すると急激に散乱強度が増加するが、24時間と72時間ではその差はわずかであった。散乱強度は樹脂の体積とポアの体積比に依存し、体積比1:1で最も増大すると考えると、4時間から24時間にかけてポア体積が急増し、その後は体積の増加は緩やかもしくは一定であると考えられる。
これらの結果から氷水中での微結晶核形成過程において、非晶質状態の繊維内部に微小な結晶核が形成され、分子鎖がわずかに凝集することで微小なポアが等方的に形成され、時間経過に伴ってこのポアが増加する。その後に繊維を延伸することでポアも延伸され、ポア同士が連結することにより細長いマイクロサイズのポアが形成されるため、微結晶核形成時間の増加に伴ってポアがより細長くなるものと考察できた。微結晶核形成時間の増加に伴うポアの細化は、これまでのSEMやX線トモグラフィー測定の結果とも一致している。非晶質繊維中の微小なポアの量を変化させた後に延伸を行うことによって、ポア形状を制御できることが見出された。
今後の課題:
今回の超小角測定によって、微結晶核形成時間の増加に伴う繊維内部のポアの増加やポア形状の細化に起因する散乱強度やストリーク散乱幅の増大を観察することができた。これまで延伸繊維のマイクロサイズのポアについてはX線トモグラフィー測定やSEMで定量できていたが、未延伸繊維中に微結晶核形成時間の増加に伴う微小なポアの増加については観測が困難であったため、超小角測定により微小なポアのサイズや量を定量できる可能性が示されたことは意義深い。
今後は微結晶核形成時間と共に微結晶核形成温度を制御することで繊維の内部構造に及ぼす影響を詳細に調べ、ポア形成機構の解明を目指すと共に繊維の力学物性との相関についても解析を行う予定である。
高分子材料の力学物性や機能は分子構造だけでなく、結晶構造、ラメラ周期、ミクロフィブリル構造等のナノ〜マイクロサイズに渡る階層構造と密接な関係があり、階層構造の精密制御が力学特性の向上に重要である。そこでマイクロサイズのポアやミクロフィブリル構造のようにナノ〜マイクロレベルに渡る階層構造を広角・小角・超小角X線散乱測定を行うことで精密に解析し、力学物性・機能と構造の相関を詳細に調べることが材料開発に重要である。
微生物産生ポリエステルのポーラス繊維は高強度かつ軽量な繊維や、内部の空隙による保温効果を有する高機能繊維としての応用が考えられている。また、ポア内に抗菌物質などの機能性薬剤を含浸し、生分解に伴う徐放能を有する多機能材料としての応用も検討している。本研究で得られた知見や測定技術は、繊維内部のポーラス構造を自在に制御できる延伸技術の開発や更なる高機能・多機能繊維の開発および評価に有用な指針を与えるものと期待できる。
参考文献:
[1] T. Tanaka, M. Fujita, A. Takeuchi, Y. Suzuki, K. Uesugi, K. Ito, T. Fujisawa, Y. Doi, and T. Iwata : Macromolecules 39,2940 (2006)
[2] T. Tanaka, K. Uesugi, A. Takeuchi, Y. Suzuki, and T. Iwata : Polymer 48, 6145 (2007)
[3] A P Hammersley, ESRF Internal Report, ESRF97HA02T, (1997)
©JASRI
(Received: April 6, 2012; Accepted: June 20, 2012; Published: February 28, 2013)
