





Volume5 No.2
SPring-8 Section A: Scientific Research Report
光電子-光イオン-光イオン同時測定法による炭酸分子の軟X線吸収スペクトル測定
The Measurement of Photoabsorption Spectra of Carbonic Acid by Photoelectron-Photoion-Photoion Coincidence Technique
a兵庫県立大学大学院物質理学研究科,b(公財)高輝度光科学研究センター
aUniversity of Hyogo, bJASRI
- Abstract
-
炭酸分子の酸素1s、炭素1sしきいエネルギー領域の吸収スペクトル測定を光電子-光イオン-光イオン同時測定法を利用して行った。得られたスペクトルには1s→π*由来と考えられるピークがあらわれたが、水および二酸化炭素の単体分子からのシグナルによる寄与がきわめて大きく、炭酸分子由来のシグナル強度も弱かったため、明確なピーク位置は確定することはできなかった。
キーワード: 炭酸、コインシデンス分光法、軟X線吸収
背景と研究目的:
二酸化炭素(CO2)は水溶液中では、(1)そのまま溶存ガスとして存在する、(2)炭酸(H2CO3)となる、(3)炭酸水素イオン(HCO3-)・炭酸イオン(CO32-)に変化する、など様々な分子またはイオンとして存在する。これらの分子およびイオンは、食品(炭酸飲料)や我々の血液中のpH緩衝系として欠かすことのできない身近な物質であり、非常に重要な生体内物質である[1-3]。
炭酸(H2CO3)は、酸性水溶液中で二酸化炭素と平衡状態で存在している。ただし、酸性水溶液中でも、その平衡が二酸化炭素+水の方に傾いているため(平衡定数Kh = 0.0037)、水溶液中では大部分は二酸化炭素として存在する。一方、アルカリ性水溶液中では、ほぼ全量が炭酸イオン、炭酸水素イオンとなり、炭酸はほとんど存在しない。また、炭酸は、通常水の中でしか存在しない。
これらの複数の要因のため、炭酸の電子状態測定は、マトリックス法などに限られていた。特に、水中でしか存在しないため、軟X線領域であるC端やO端での吸収スペクトルつまりXAFS測定には制限があり、これまで測定されることはなかった[4, 5]。
そこで我々は、光電子-光イオン-光イオン同時測定法(以後PEPIPICO法)[6]により、軟X線領域での炭酸分子の吸収スペクトル測定を試み、炭酸の直接吸収スペクトル測定を行ったので報告する。
本研究の特色にはいろいろあるが、まずクラスタービーム生成により、炭酸分子を真空中に生成したことである。クラスタービーム法は、数気圧の背圧のもとで分子を真空中に噴出することにより、低温状態で原子や分子を結合させるものである。ヘリウム希釈した水と二酸化炭素を真空チャンバーに噴出させることにより、水中でなくとも炭酸分子を取り出すことができる。
さて、通常のクラスター実験では、軟X線を照射し生成したイオンのイオン収量により吸収スペクトルとする。しかし、内殻励起では、ほとんどの分子はイオン化解離をしてしまい解離せず残るものはわずかである。また、炭酸分子の場合、不安定であるために解離しやすく、ほぼ全部がイオン化解離する。さらに、本実験条件の場合、水と二酸化炭素が大量に含まれているため、生成したイオンが、水または二酸化炭素から生成したものと区別することができない。
そこで、PEPIPICO法により、親分子を特定しながら吸収スペクトル測定を行った。PEPIPICO法とは飛行時間型質量分析器をつかい、飛行時間によりイオン種を選別するものである。生成する電子のシグナルをスタート信号として、生成したイオン分子、またはイオンフラグメントから複数のイオンの飛行時間を測定する。この時、二体解離であれば、2つのイオンを足し合わせることで親イオンが特定できる。例えば、炭酸分子であれば OH++HCO++O のような解離チャンネルを特定することで、炭酸を親分子として再現することができる。
この方法の特徴は、バックグランドシグナルを除くことができる点にある。例えば、本実験では、水分子と二酸化炭素分子も共存するため、そこから生成する水素イオン、酸素イオン、ラジカルイオンが、実際の炭酸分子由来によるシグナルより圧倒的に強い。そこで、2つの解離イオンの相関を二次元スペクトルとし、この二次元スペクトルの一部分を選択することで、水や二酸化炭素由来である、H++OH+ や C++CO+ などのチャネルを画像上の別点として取り除くことができる。その結果、S/N比を向上させることができる。これは、まさに、少量しか生成していない炭酸分子を測定する方法としては最適である。
また、この方法のユニークな点は解離チャネルを様々に選べる点にある。例えば OH++HCO2+ などといった解離チャネルが観測された場合は、解離の時間スケールで水素原子が分子内移動していることを示唆するので、化学反応という観点から非常に興味深い。
実験:
実験はSPring-8のBL27SUで行った。上に記したように、溶液中では炭酸の吸収スペクトル測定は、多量の二酸化炭素の存在のためむずかしい。そこで、ヘリウム希釈した10%の二酸化炭素ガスを重水(D2O)中でバブリングし、背圧1気圧で真空チェンバーに噴射し、クラスター状態にして軟X線を照射した。炭酸の生成は四重極質量分析装置により、質量数64(D2CO3)の生成により確認した。
実験では、生成した電子とイオンを同時測定法により検出した[6]。軟X線を照射し放出した電子が検出器に到達した時間を0とする。次に飛行時間型質量分析器により、一つ目のイオンが検出器に到着した時間をT1、二つ目のイオンが到着した時間をT2とする。T1とT2の二次元スペクトル(PEPIPICOスペクトル)の一部分を切り出すことで、特定の生成イオン対を選択することができる。図1に示したのは、すべての光エネルギーでのスペクトルを足し合わせたときのPEPIPICOスペクトルである。なお、およそひとつのエネルギーあたり10分程度を積算した。
実験では炭酸分子からのみ生成すると考えられる、OD++DCO+、D++CO3+、OD++DCO2+ など、まず特定の2つのイオン生成を選択した2次元スペクトル解析を行った。そして、この図の一部分を切り出して、光エネルギーごとに積算したものを部分イオン対生成収量エネルギースペクトルとした。最終的に光エネルギーに対してバックグランドを差し引きながらプロットすることで、炭酸のみの軟X線吸収スペクトル測定が可能となる。
酸素1sのイオン化しきいエネルギー領域(532 eV ~ 542 eV)と炭素1sのイオン化しきいエネルギー領域(288 eV ~ 298 eV)の2つの領域で0.05 eVきざみで201枚のPEPIPICO二次元マップを解析して、部分イオン対生成収量スペクトルを作成した。
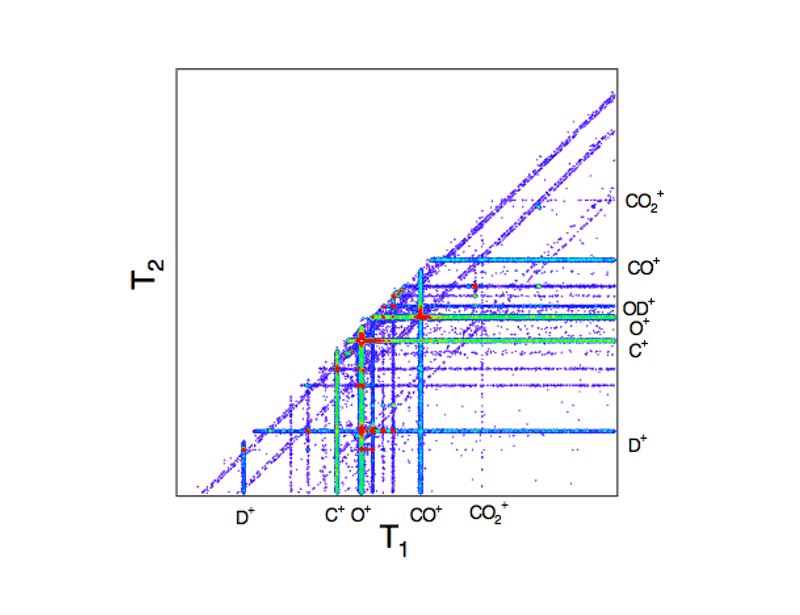
図1. 一つ目のイオンが検出器に到着した時間をT1、二つ目のイオンが到着した時間をT2とした時のT1とT2の二次元スペクトル。白色は強度が強いことを表す。
結果および考察:
図1に、縦軸に最初に検出器に到達したイオンの飛行時間T1 、横軸に次のイオンの飛行時間T2の2次元マップを示した。図に示すように、二酸化炭素、水の単体分子から生成したシグナルが非常に強く、炭酸から生成した部分はシグナルが弱い。そこで、炭酸由来の部分のみを抽出して、バックグランドを補正して吸収スペクトルを求めた。
図2に、PEPICO法によって求めたO1s付近の生成イオン収量スペクトルを2つ(D++CO3+、OD++DCO2+)、C1s付近の生成イオン対収量スペクトルひとつ(OD++DCO+)を示す。求めた吸収スペクトルには酸素および炭素の1s-π*によるピークが観測された。理論計算では、炭素の1s-π*によるピークは290 eV付近と予想され、その位置に非常に近い。しかし、炭酸分子由来のシグナル強度が弱かったため、明確なピーク位置は確定することはできなかった。


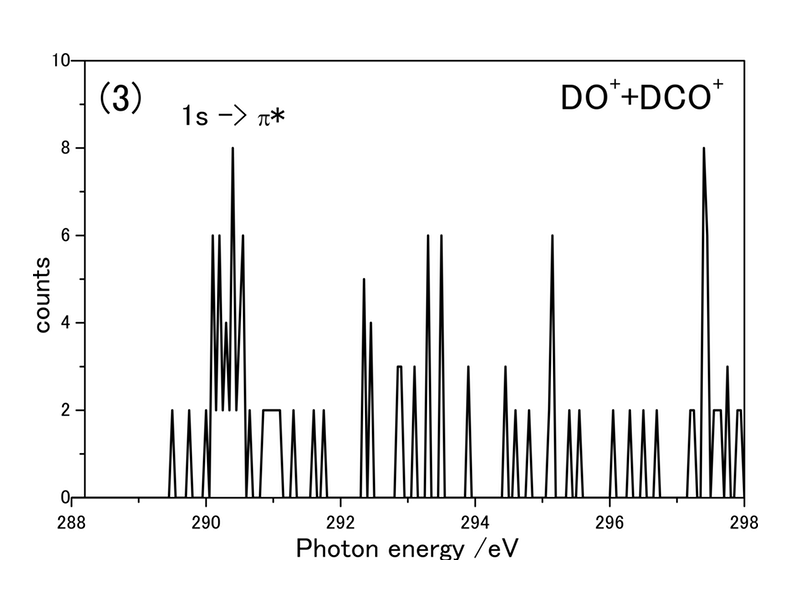
図2. PEPICO法によって求めた(1)D++CO3+(O1s付近)(2)DO++DCO2+(O1s付近)(3)DO++DCO+(C1s付近)、生成イオン対収量スペクトル
今後の課題:
炭酸分子の酸素1s、炭素1sしきいエネルギー領域の吸収スペクトル測定を光電子-光イオン-光イオン同時測定法を利用して行った。得られたスペクトルには1s→π*由来と考えられるピークがあらわれたが、水および二酸化炭素の単体分子からのシグナルによる寄与がきわめて大きく、炭酸分子由来のシグナル強度も弱かったため、明確なピーク位置は確定することはできなかった。
今回の最大の問題点は水、二酸化炭素に対して、圧倒的に炭酸分子が少なかったためと考えられる。通常は、放電により生成量を高めることができるが、この方法はイオンが発生するため、シグナルが著しく毀損するという問題点がある。これらの問題解決が課題である。
参考文献:
[1] T. Kokido, H. Sangyo, Chem. Phys. Rev., 104, 4891 (2004).
[2] P. Falkowski et al, Science, 290, 291 (2000).
[3] K. Caldeira, M.E. Wickett, Nature, 425, 365 (2003).
[4] A. H. England et al, Chem. Phys. Let., 514, 187 (2011).
[5] Y. Horikawa et al, Journal of Molecular Liquids, 189, 9 (2014).
[6] Y. Tamenori et al, J. Chem. Phys., 134, 204302 (2011).
ⒸJASRI
(Received: March 31, 2017; Early edition: July 11, 2017; Accepted: July 18, 2017; Published: August 17, 2017)
