





Volume5 No.2
SPring-8 Section A: Scientific Research Report
溶媒誘起結晶化現象を利用したPETボトルのエコ製造のための構造化学的根拠の探究
Structural Study of Solvent-induced Crystallization Phenomenon of PET for the Economical Production of PET Bottles
豊田工業大学
Toyota Technological Institute
- Abstract
-
非晶性PET試料の溶媒誘起結晶化過程における階層構造発展過程を透過赤外スペクトル、広角X線回折および小角X線散乱同時・時間分解測定によって追跡し、結晶格子の形成ならびにラメラ積層構造の形成の様子を詳細に調べた。
キーワード: ポリエチレンテレフタラート、溶媒誘起結晶化現象、X線散乱、赤外分光
背景と研究目的:
PETボトルは我々の生活の中に深く定着している。従来の製造法では、試験管の形をしたPETチューブ(プリフォーム)を加熱金属枠の中に入れて金属棒と圧縮空気で変形させ、ガラス転移点以上で一定時間熱処理をして作成する。熱処理したボトルは、沸騰水を入れても変形しないので、熱いお茶用などに使う。このプロセスでは相当量の熱エネルギーを利用し、しかも熱処理に長い時間を要する。石鍋らは、PETのプリフォームをあらかじめアセトアルデヒドやトルエンに短時間浸漬させた後に圧縮空気で成形すると、比較的低温で、しかも僅かな熱処理時間で熱湯にも耐え得るボトルの出来上がることを報告した[1, 2]。その構造化学的理由を探るべく、溶媒に浸漬したプリフォームの赤外スペクトルを測定したところ、加熱前にも拘わらずPETの結晶性バンドが強く検出された。つまり溶媒誘起結晶化がプリフォームの段階で生じている。比較的低温で結晶化度を保ったまま成形する結果として、高温での熱処理を特に行うこともなく高耐熱性ボトルが出来上がったと推定される。しかし、それだけでは、如何なる機構で溶媒誘起結晶化が生じているのかが不明で、どのような高次構造の形成が耐熱性を賦与させているのかも分からない。
溶媒誘起結晶化はPET以外にも、シンジオタクティックポリスチレンなど様々のポリマーについて知られた現象であるが、結晶化機構を詳しく調べた例はあまり多くない[3]。溶媒結晶化における高次構造の発達など微視的観点から詳しく解明することで、溶媒分子の役割が見える。本研究では、溶媒蒸気雰囲気下で誘起されるPET試料内部での構造変化を、赤外スペクトル、広角(WAXD)及び小角X線散乱(SAXS)の同時・高速時間分解測定によって追跡した。PETについてWAXD、SAXS測定に基づく溶媒誘起結晶化の論文は意外と限られており[4-6]、本実験のように、PET試料の溶媒誘起結晶化過程について分子鎖レベルから結晶、非晶相中での分子鎖集合状態さらにはナノスケールの高次組織まで、幅広い階層構造の変化を一度に、しかも時間の関数として追跡することに基づく検討は、これまでに報告されたことはない。
実験:
実験はSPring-8のBL40B2で行った。Figure 1の配置で、透過型赤外分光器(携帯型FTIR Alpha、Bruker社製)、WAXD用検出器およびSAXS用検出器を設置し、赤外スペクトル、WAXDおよびSAXS二次元像の同時・時間分解測定を行った[7]。WAXD用検出器としてはフラットパネルセンサ(C9728-DK、浜松ホトニクス社製)、SAXS用検出器にはX線イメージングインテンシファイア(V7735P、浜松ホトニクス社製)付きCCDカメラ(C4742-98、浜松ホトニクス社製)を用いた。試料としては市販のPETペレットを100℃以上で一晩、真空乾燥させたものを用い、溶融後、液体窒素温度に急冷した非晶性フィルムを実験に供した。このフィルムを室温で約4倍延伸し、配向試料を作成した。フィルムの厚さは、無配向試料で凡そ150 μm、配向試料で290 μmであった。これらの試料を、溶媒溜めをもった自家製セルにセットした。セルの窓には剥離した雲母薄膜を使用した。セルの底から溶媒を注入し溶媒蒸気雰囲気に試料フィルムを暴露し、その後のWAXD、SAXSおよび赤外スペクトルの変化を一定時間間隔ごとに測定していった。測定は室温で行った。PETの良溶媒として用いたヘキサフルオロイソプロパノール(HFIP)の場合、結晶化は速く起こったが、短時間の間にフィルムは溶解してしまった。そこで、クロロフォルムをHFIP/CHCl3体積比率1/3~1/15で混合させたものを溶媒として用い、溶媒結晶化速度を調節した。X線の測定時間間隔はWAXDが10秒、SAXSが1~2分、赤外は32秒であった。

Figure 1. Setting of sample cell on the stage.
結果および考察:
(1)無配向試料(室温)
無配向非晶性PETフィルム(厚さ150 μm)をHFIP/CHCl3(比率6/94%)の混合溶媒蒸気中に暴露し、WAXDおよびSAXS 2次元パターンの時間変化を測定した。この2次元データを円環積分し1次元プロファイルに直したものがFigure 2である。WAXDプロファイルは数多くの結晶ピークと非晶ピークが複雑に重なっているために、一連のプロファイルのピーク分離を系統的に解析することが必要である。ここでは、まず、出発の非晶状態および実験最終時の部分結晶状態のプロファイルを波形分離し、その結果を基本として結晶化途中のプロファイルを処理した(各ピーク成分の位置と幅を固定)。そして全体の面積強度に対する結晶性ピーク面積強度の比(Xc)を結晶化度の目安とした。SAXSデータ解析はバックグラウンド補正後のプロファイルを、積層ラメラ相関関数を用いて解析し、ラメラ長周期などを見積もった。Figure 3に、解析結果をまとめて示す。溶媒注入(時間0)から約9分経過後に結晶性WAXDピークが出現しはじめ、平行して積層ラメラ特有の長周期ピークもSAXSに現れ始めた。その後の長周期(LP)およびラメラ平均厚さ<d>の変化は僅かであるが、インバリアントQはWAXDのXcと似た増加を示しており、ラメラの数が次第に増加していることがわかる。Figure 4は赤外スペクトルの時間変化である。熱処理を施したPETフィルムのスペクトルも参考に載せた(破線)。主にメチレン基の振動に関するバンドが結晶性と非晶性とで明確に区別できる。2909 cm-1バンドは結晶性メチレン伸縮モード[v(CH2)]に対応し、X線と同じタイミングで強度を増加させていった。他に1471 cm-1 [CH2はさみ振動], 1384 cm-1 [CH2縦揺れ振動]も結晶化に伴って強度を増していった。ベンゼン環に関わるバンドの変化は殆どなかった。3024 cm-1はクロロフォルム液体のバンド(3019 cm-1)、3431 cm-1はHFIP液体のバンドに近く、試料セルの窓に凝結した溶媒液体によるのかもしれない。クロロフォルムのバンドのシフトを考えると試料に吸収されたクロロフォルムに基づいているとも考えられるが、今後検討したい。
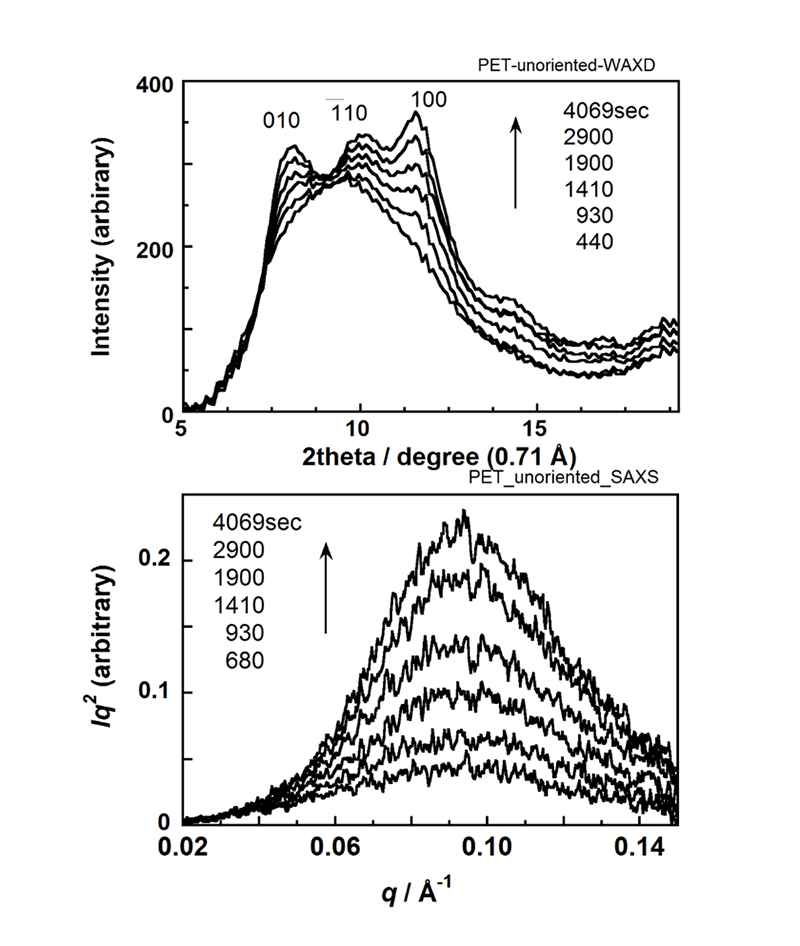
Figure 2. Time dependences of (a) WAXD and (b) SAXS profiles in the solvent-induced crystallization process of PET film.
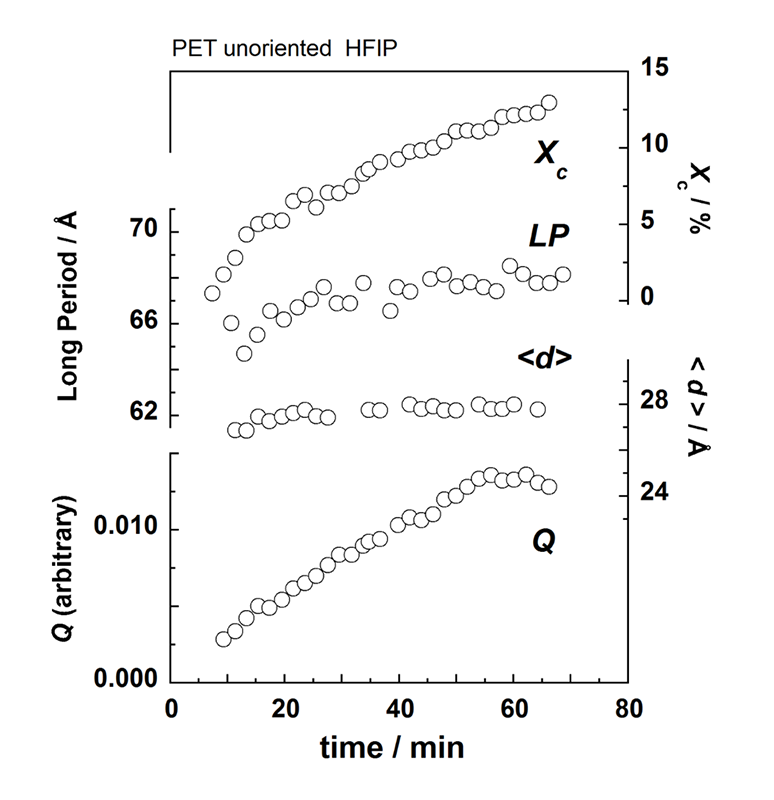
Figure 3. Time dependence of the relative crystallinity (Xc), the long period (LP), the average lamellar thickness <d>, and invariant Q estimated from Figure 2.
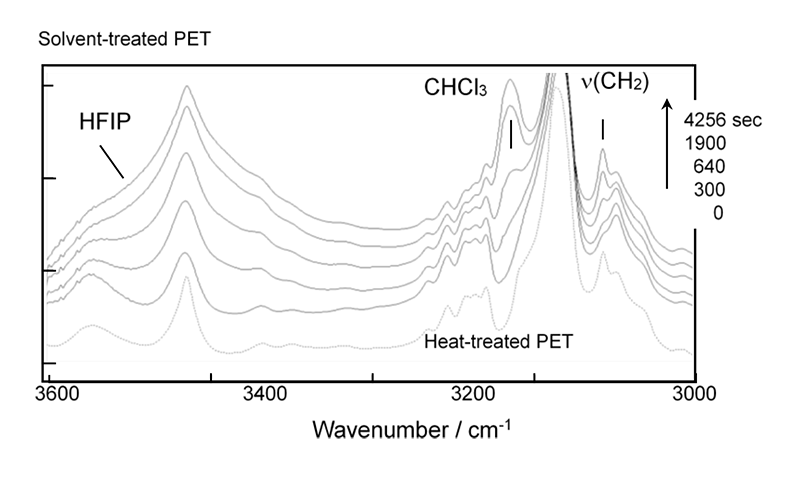
Figure 4. Time dependence of IR spectra measured during the solvent-induced crystallization process of the unoriented amorphous PET film.
(2)配向試料
Figure 5に、1軸配向フィルム (厚さ290 μm)を室温においてHFIP/クロロフォルム蒸気に曝したときのWAXDおよびSAXS 2次元パターンの時間変化を示す。出発試料は、いわゆるメゾ相(中間相)である。メゾ相は、コンフォーメーションの乱れた分子鎖が延伸方向に配向した。しかし隣接分子鎖間の相対的な高さにあまり強い相関のないネマティック液晶的集合体と考えられている[8]。WAXDパターンについては、溶媒の侵入とともに(010)、(110)、(100)など結晶性反射が次第に出現し、強度を増していった。SAXSパターンについても、出発時の試料ではラメラ積層構造に基づくピークは観測されず、メゾ相と非晶相との密度に大きな差が見られないことが原因している。結晶化が始まると4点散乱像が初めはぼんやりと、そして次第に明瞭になっていった。
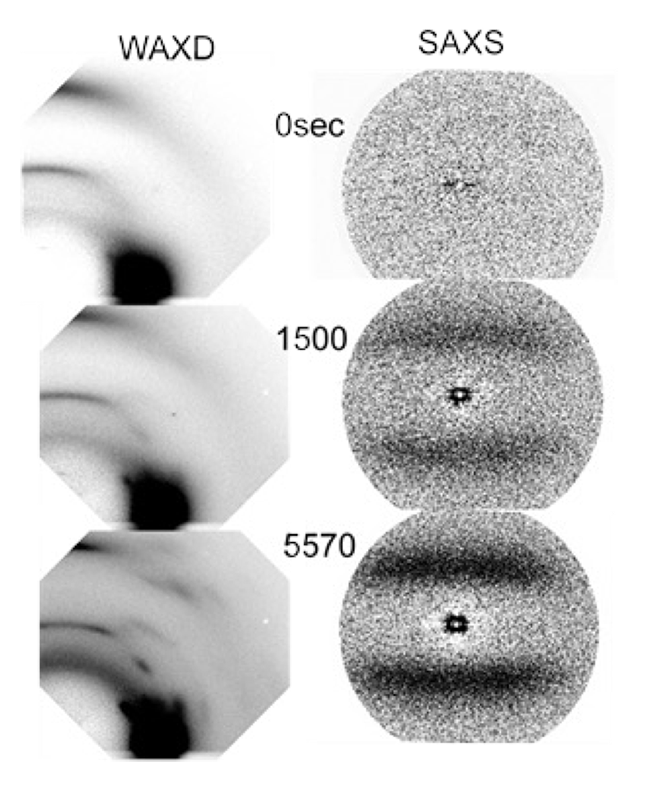
Figure 5. Time dependence of 2D-WAXD and SAXS patterns in the solvent-induced crystallization of an oriented PET film.
4点散乱像の解析から、ラメラの積層構造についてはFigure 6のような変化が見出された(SAXSのデータ解析精度は比較的低いが、構造変化の傾向は把握できる)。Figure 7に構造発展のイメージを示す。溶媒侵入でミクロブラウン運動が活発になった分子鎖は、横方向に規則化し、メゾ相から結晶相に変化する。ラメラは傾斜している。時間とともに、より安定な充填をするように分子鎖は軸方向にスリップし、結果的にラメラ表面の傾きが増えると考えられる(Figure 6におけるラメラ傾斜角度の増大)。
Figure 3の無配向試料とFigure 6の配向試料について両者の結晶化度の成長の様子を比較すると、無配向試料では溶媒注入ののち10分ほど経ってから結晶化が始まったのに対し、配向試料では素早く開始している。配向試料の方が2倍も厚い、つまり溶媒の浸透拡散に長い時間が要るにも拘わらず結晶化が早く始まっているのは、Figure 7のように分子鎖が非晶中でもある程度配向しており規則的構造への移行が容易に起こるためと考えられる。「背景」で紹介した石鍋らのPETボトル作成法における「メルト成形したプリフォームに短時間、溶媒浸漬した場合に結晶性の比較的高い試料が得られる」との内容について、射出成形して作られるプリフォーム中での分子鎖配向を制御することでさらに効率の良いプロセスに改良出来ることがFigure 7から示唆される。
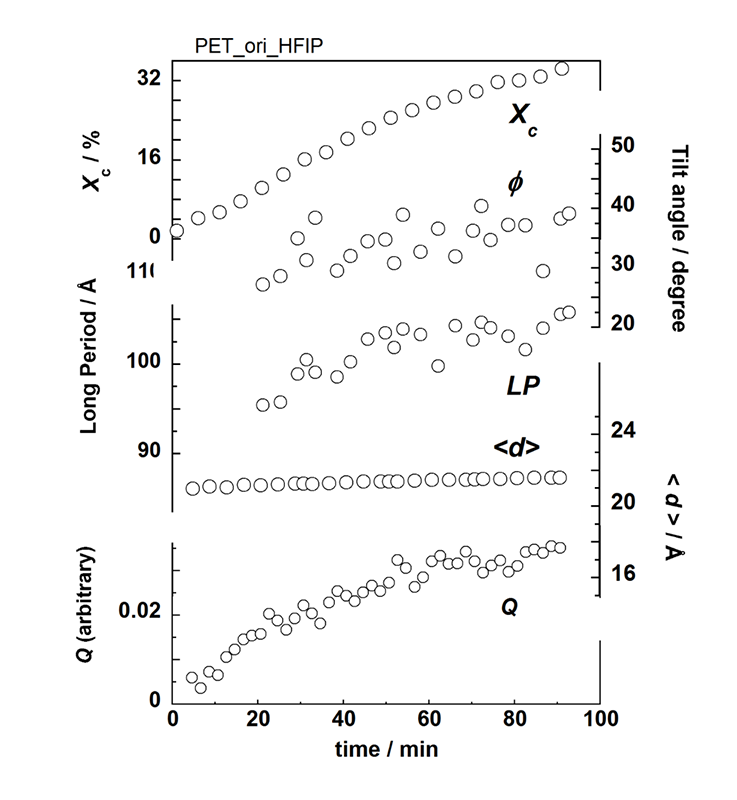
Figure 6. Time dependence of the relative crystallinity Xc, the lamellar tilting angle φ, the long period LP, the averaged lamellar thickness <d>, and the invariant Q estimated from the data analysis of Figure 5.

Figure 7. Schematic illustration of the solvent-induced crystallization of oriented PET sample. (a) oriented meso phase, (b) start of micro-Brownian motion, and (c) formation of tilted lamellar structure.
今後の課題:
以上のように、PET非晶試料の溶媒誘起結晶化現象における階層構造変化を追跡した。予備的な実験に終わってしまった感が強いが、試料周りの工夫や検出器感度などの改善によって、階層構造の発展過程をより詳細に解明できると考えている。本実験の経験を基に、今後解決すべき事項を並べてみた。
① 有機溶媒として、当初の目的では参考文献[1]で用いたトルエンやアセトアルデヒドを試みる予定であったが、今回の実験では極性の高いHFIPを溶媒として用いた。トルエンやアセトアルデヒドによる溶媒誘起結晶化が非常にゆっくりとしか起こらないためである。しかしHFIPのように異なる溶媒を用いても、結晶化に際して生じている構造発展過程は本質的に同じであると考えている。シンジオタクティックポリスチレンで議論したような[3]溶媒分子の種類に依存する結晶化速度の差異の検出、そして、それについての運動学的議論を行うには種々の溶媒を用いる必要があり、これは今後の課題である。
② 今回、赤外スペクトル、WAXDおよびSAXSの同時測定・時間分解測定実験を行ったが、フィルムが厚かったために、赤外スペクトルについては、解析可能な波数領域が3000 cm-1付近に限定されてしまった。試料部分の厚みを場所によって変え、赤外測定用の箇所は薄いフィルムを、X線散乱測定箇所は薄いフィルムを重ねて用いる、というようにすることで、この問題はクリアできると思われるが、両方の箇所において溶媒が同じ拡散速度で浸透していくことが保証されていなければならない。
参考文献:
[1] M. Ishinabe, K. Tashiro, K. Imaeda, D. Yamamoto, Nippon Kikai Gakkai Ronbunshu A, 77, 526 (2011).
[2] M. Ishinabe, K. Tashiro, K. Imaeda, Y. Yamashita, K. Hayashi, Nippon Kikai Gakkai Ronbunshu A, 78, 81(2012).
[3] E. Bhoje Gowd, K. Tashiro, C. Ramesh, Progr. Polym. Sci., 34, 280 (2009).
[4] P. J. Makarewicz, G. L. Wilkes, J. Polym. Sci., Polym. Phys. Ed., 16, 1529 (1978).
[5] A. K. Kulshreshtha, A. H. Khan G. L. Madan, Polymer, 19, 819 (1978).
[6] R. Khanum, W. Takarada, A. Aneja, T. Kikutani, Polymer, 59, 26 (2015).
[7] K. Tashiro, H. Yamamoto, T. Yoshika, T. H. Ninh, M. Tasaki, S. Shimada,T. Nakatani, H. Iwamoto, N. Ohta, H. Masunaga, Macromolecules, 47, 2052 (2014).
[8] F. Auriemma, C. De Rosa, P. Corradini, Avd. Polym. Sci., 181, 1 (2005).
ⒸJASRI
(Received: March 11, 2017; Early edition: July 11, 2017; Accepted: July 18, 2017; Published: August 17, 2017)
