





Volume5 No.1
SPring-8 Section A: Scientific Research Report
Evaluation by HAXPES of the Chemical State of the Components of a Close-packed Array of Bi-metallic (Au-Ag) Nanoparticles on ITO
a National Institute for Material Science, b SPring-8 Service Co., Ltd
- Abstract
-
Hard X-ray Photoemission Spectroscopy was employed to investigate the elemental composition and chemical state of core-shell bimetallic nanoparticles deposited on a solid substrate. A shift in Au 4f7/2 and Ag 3d5/2 binding energies together with the change in surface elemental composition of the nanoparticles suggests that a low-temperature alloying occurs for bimetallic nanoparticles.
Keywords: metallic nanoparticles, core shell nanoparticle, plasmonics, photoemission spectroscopy
Background and Purpose
The development of ordered plasmonic substrates comprising complex metallic structures with tunable optical properties is of particular interest nowadays, due to their promising applications in multiple fields, such as photovoltaic [1], biology [2], chemistry [3,4], and environmental science [5]. Bottom-up deposition processes have gained huge attention recently for their ability to obtain large scale and cheap plasmonic substrates without compromising their uniformity and optical response [6,7]. Nevertheless, bottom-up plasmonic substrates can suffer from some critical limitations compared to top-down (e.g. lithography-based) arrays, which are linked to the limited chemical and mechanical stability of the supported nanoparticles (NPs) to the harsh conditions required for many real life applications, such as high temperature, presence of strong oxidizers (e.g. catalysis) or repeated wash cycles (e.g. biosensing). A novel bottom-up deposition method to obtain two dimensional arrays of metallic nanoparticle on a solid substrate has recently been developed in our laboratory [7,8], combining three deposition methods, namely electrophoresis, solvent evaporation and self-assembly. This deposition method is very flexible, since it allows the deposition of NPs of different size, shape and elemental composition. Recently, our attention has been focused on the preparation of two dimensional arrays of core-shell bimetallic NPs, containing silver and gold, synthesized by wet chemical processes. The NPs are prepared by a step-wise procedure and later chemically immobilized on an indium-tin oxide (ITO) thin film on a quartz substrate for their use as plasmonic solid substrate.
The purpose of our experiment was to verify the chemical state of the NPs after deposition on the substrate and thermal processing. Nanoparticles, in fact, are susceptible to oxidization or alloying during the functionalization, deposition and thermal treatment steps, and this would strongly affect their plasmonic properties. When oxidized, metals lose their plasmonic properties, therefore it is of the upmost importance to minimize the oxidation of the metals (especially silver, in our case). Several reports have revealed that the presence of an electron transfer effect at the interface between a gold core and a thin silver shell might increase the chemical stability of silver, inhibiting the galvanic replacement reaction with gold ions in solution and reducing silver oxidation [9–11]. As a result, due to their tunable optical properties and improved chemical stability, gold core silver shell NPs have recently been identified as optimal candidates for the replacement of pure silver NPs for plasmonic or surface-enhanced Raman scattering applications. Nevertheless, the improved chemical stability of the silver shell has been attributed to an interface phenomenon, therefore this property might be susceptible to the atomic diffusion between the two metals and to the subsequent alloying of the NPs. Recently, several reports have suggested that many properties of NPs are size-dependent and consequently differ remarkably from their bulk counterparts [12], including the ability to form alloys [13]. In fact, alloying of small gold core silver shell NPs has been reported to occur at very mild temperature conditions, with estimated atomic diffusion coefficients that are orders of magnitude higher than for bulk materials [13]. Furthermore, the crystalline state of the NPs has also been reported to be affected by low temperature annealing, with temperatures as low as 120 °C [14]. In addition to the size, surfactants and supporting materials have also been found to affect the ability of bimetallic NPs to form alloys or control the crystalline phase [15]. Therefore, it is of outmost importance to characterize the chemical state of the NPs on the substrate, since silver oxidation and alloying during the synthesis, electrodeposition and annealing steps might affect the optical and chemical properties of the NPs array. For this purpose, we employed photoelectron spectroscopy, in particular hard X-ray photoelectron spectroscopy, to study the chemical state of the NPs. Samples were also annealed at different temperatures to evaluate the possible change in the crystalline structure or alloying.
Experimental Summary
Nanoparticles synthesis
Gold core silver shell NPs have been synthesized by a multi-step synthetic procedure [8], unlike the previously reported one-pot synthesis [9]. Freshly prepared NPs were characterized by transmission electron microscopy (TEM) and scanning electron microscopy (SEM) to determine their size and shape. Gold NPs have been synthesized by a two-step synthetic protocol, at first 13 nm spherical gold nanoparticles (AuNPs) have been obtained by a standard citrate reduction method [16], then the 13 nm AuNPs have been used as seed for the growth of larger AuNPs (30.0 nm in diameter) [17]. This second step involved the use of a small amount of silver nitrate (4 at% final Ag content homogeneously distributed in the topmost 8.5 nm of the large AuNPs, no core-shell structure could be identified) to maintain the spherical shape and narrow size distribution of the NPs. After the large gold cores have been synthesized, the silver shell was deposited with ascorbic-acid as room-temperature reducing agent according to literature [18]. The final diameter of the spherical gold core silver shell nanoparticles (Au@AgNPs) was found to be 36.0 nm. Finally, double shell particles (Au@Ag@AuNPs) with an additional thin gold shell (0.2 nm) were synthesized by adding chloroauric acid and sodium citrate to the Au@AgNPs while continuously refluxing for 30 minutes at 100 °C (final diameter 36.4 nm). The deposition of a silver shell and a second gold shell was confirmed by TEM and energy-dispersive X-ray diffraction (EDX), suggesting that our synthetic method, although different from the one-pot synthetic method previously reported in literature [11], can guarantee the synthesis of double shell particles. In addition to these NPs, 20 nm silver nanoparticles (AgNPs) were synthesized according to the procedure reported in literature [19], and commercially available 50 nm AuNPs were purchased from BBI International and used as reference standards. Afterwards, all NPs were functionalized with mixed alkanethiols according to our previously published method [3,8,20,21].
Nanoparticles deposition
Alkanethiol-capped NPs were then deposited on an ITO coated quartz substrate (ITO 300 nm /quartz 635 μm). The ITO/quartz substrate was functionalized with a mixed monolayer of hexanedithiols (Hexdt) and mercaptopropyltrimethoxysilane (MPTMS) by a successive immersion in a Hexdt solution 5% w/v in ethanol and in a MPTMS 5% in toluene. The sulfur-terminated substrate was then used as an anode for the electrodeposition of alkanethiol-capped metallic NPs, according to a protocol developed at our laboratory [7]. This deposition method combines electrodeposition to solvent evaporation and self-assembly. The alkanethiol-capped metallic NPs were redispersed in a solution of hexane:acetone 9:1 and the sulfur-terminated ITO substrate was immersed in the solution. A constant voltage (1 V DC) was applied between the ITO substrate and a plastic carbon counter-electrode to allow for electrodeposition of the positively charged metallic NPs. The solvent was left to evaporate in a nitrogen-filled glove bag; this process allowed for the rearrangement of the NPs at the air-water interface to form a hexagonally close-packed array of NPs. After the complete evaporation of the organic solvent, the quartz/ITO substrate coated with metallic NPs was annealed at 60 °C overnight (on a hot plate in a capped container) to allow for the formation of a covalent bond between the terminal thiol group of the substrate and the metallic NPs (sulfur-metal bond). To evaluate the effect of annealing temperature, a batch of samples was annealed at 120 °C overnight. The substrates were then sonicated to remove physisorbed NPs and stored in a nitrogen purged environment.
Nanoparticles characterization
In order to verify the morphology and elemental composition of the NPs arrays, the samples were characterized by SEM without any further treatment. The NPs surface density was estimated from SEM to be of the order of 1012 NPs/cm2. In addition to SEM characterization, the sample’s lateral cross section was obtained by focused ion beam (FIB) milling and TEM and EDX analysis. The sample preparation procedure (FIB milling) to observe the lateral cross section, nevertheless, is likely to introduce contaminants into the sample, therefore no final conclusion could be made about the elemental composition and layered structure of the NPs, i.e. whether alloying or oxidation took place during the deposition and annealing process. For this reason, it was necessary to investigate the sample using a non-destructive technique. X-ray photoelectron spectroscopy data were therefore acquired, nevertheless, it was difficult to collect information about the core material for core-shell and double shell NPs possessing thick shell layers. Different ITO thicknesses and conductive coating (amorphous carbon layers of various thicknesses were deposited on the samples) were tested in order to find the optimal conditions and to avoid surface charging.
Hard X-ray Photoelectron Spectroscopy
For our experiment at BL15XU beamline at SPring-8 we employed a 5.95 keV energy hard X-ray source, total energy resolution of 238 meV, passing energy of 200 eV (unless otherwise stated), a take-off angle of 88 °, room temperature and pressure of about 3 × 10-7 Pa. In our experiment, we analyzed the following binding energies: Au 4f, Ag 3d, valence band structure, C 1s, S 1s and O 1s. The Au 4f, Ag 3d, C 1s, O 1s and S 1s XPS spectra were analyzed with CASA XPS, a Shirley baseline was applied and the peaks were fitted with a product Gaussian-Lorentzian lineshape. The Au 4f7/2 level was used for binding energy (BE) calibration and Au Fermi energy for the calculation of the total energy resolution.
Results and Discussion
The Au 4f and Ag 3d HAXPES spectra were collected for all samples and the Au 4f7/2 and Ag 3d5/2 were shown in Fig. 1. All spectra in Fig. 1 were fitted with a single Gaussian-Lorentzian peak (80% Lorentzian, 20% Gaussian contribution) and the resulting peak positions were found to correspond to the typical BE of metallic elements. The peak position together with the presence of the metallic Ag loss peak at 372 eV (not shown) and the lack of increase in oxygen content (data not shown) suggest that metals are present in their valence zero state with only a negligible contribution from oxidized species (silver oxide species present broader peaks at lower binding energies). Analyzing the Au 4f7/2 and Ag 3d5/2 peak position for the different samples (see Table 1 and Fig. 2), we can notice that there is a positive energy shift for the Au 4f7/2 peak for samples annealed at 60 °C with the increase in shell number, while the opposite tendency is observed for Ag 3d5/2 peaks. This tendency is similar to what has been previously reported for citrate-capped colloidal particles [3], a phenomenon attributed to the electron transfer effect that occurs at the interface between gold and silver. In our experiment, nevertheless, the Au 4f7/2 shift was found to be smaller than previously reported (shifts of 0.022 eV and 0.023 eV instead of 0.05 eV and 0.1 eV, respectively for core-shell and double shell NPs [11]). The smaller shift can be due to the fact that our NPs are bigger (with consequently smaller surface to volume ratio) than the ones previously investigated (diameter of 30-36.4 instead of previously reported 14-23 nm) and hard X-rays have larger penetrations depth. Under these experimental conditions, the surface contribution might be partially masked by the bulk contribution. More interestingly, as shown in Fig. 2, for samples annealed at 120 °C, the Au 4f7/2 BE are almost constant for all NPs. The Au 4f7/2 BE for AuNPs annealed at 120 °C shows a large and positive shift compared to the AuNPs annealed at 60 °C, while the Au 4f7/2 BE for Au@AgNPs and Au@Ag@AuNPs annealed at 120 °C are comparable although slightly lower than the BE of the same samples annealed at 60 °C.

Fig. 1. (a) HAXPES Au 4f7/2 spectra (markers are raw experimental data, solid lines are best fit). Black circles correspond to AuNP annealed at 60 °C, black triangles to Au@AgNP annealed at 60 °C, black squares to Au@Ag@AuNP annealed at 60 °C, blue circles to AuNP annealed at 120 °C, blue triangles to Au@AgNP annealed at 120 °C and blue squares to Au@Ag@AuNP annealed at 120 °C. The solid lines represent the best fit with Gaussian-Lorentzian curves. The dashed vertical line remarks the Au 4f7/2 BE for AuNPs annealed at 60 °C. (b) HAXPES Ag 3d5/2 spectra (markers are raw experimental data, solid lines best fit). Black circles correspond to AgNP annealed at 60 °C (raw data), black triangles to Au@AgNP annealed at 60 °C, black squares to Au@Ag@AuNP annealed at 60 °C, blue triangles to Au@AgNP annealed at 120 °C and blue squares to Au@Ag@AuNP annealed at 120 °C. The solid lines represent the best fit with Gaussian-Lorentzian curves. The dashed vertical line remarks the Ag 3d5/2 BE for AgNPs annealed at 60 °C.
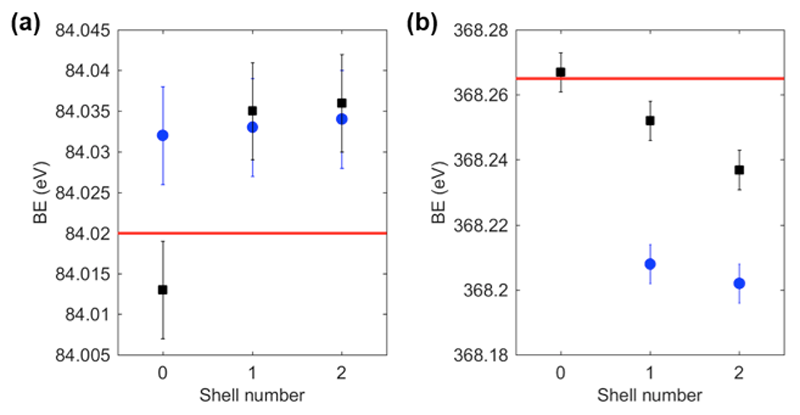
Fig. 2. (a) Au 4f7/2 BE for nanoparticles samples and reference gold foil. Black squares represent nanoparticles annealed at 60 °C, blue circles nanoparticles annealed at 120 °C, while the red line represents bulk gold. (b) Ag 3d5/2 BE for nanoparticles samples and reference silver film. Black squares represent nanoparticles annealed at 60 °C, blue circles nanoparticles annealed at 120 °C, while the red line represents bulk silver.
Table 1. Metals BE and their full width at half maxima (FWHM).
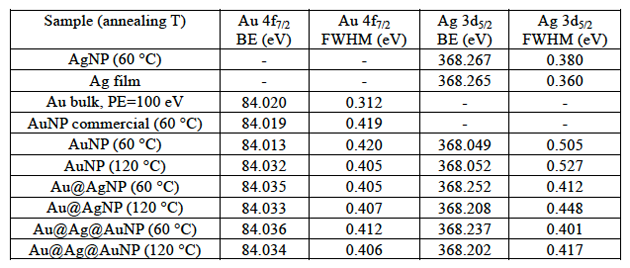
From Fig. 2 we can also notice that the Ag 3d5/2 BE for all samples annealed at 120 °C are found to be lower than the binding energies for NPs annealed at 60 °C. The origin of this negative shift can be attributed to multiple causes, most notably surface oxidation and interdiffusion of the atoms with consequent alloying. In fact, low temperature annealing of NPs on solid substrates was found to induce a wealth of phenomena such as crystallization, phase segregation and sintering of gold-platinum NPs [22], and alloying for a mixture of colloidal solutions of alkanethiol-capped gold and silver NPs [23]. In addition, for smaller gold core silver shell NPs in water solution, spontaneous alloying has been reported and attributed to the enhanced diffusion coefficient due to the high density of defects at the Au-Ag interface [13].
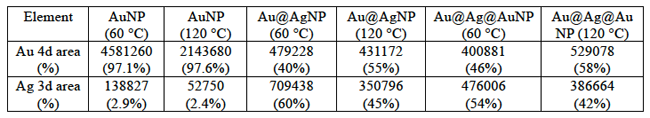
To determine the origin of the BE shifts for both Au 4f7/2 and Ag 3d5/2 peaks in samples annealed at 120 °C, we analyzed the HAXPES survey scans to estimate the elemental composition of the samples. From Fig. 3 and Table 2, we can notice that the gold content seems to increase for samples annealed at 120 °C, especially for Au@AgNPs. Since HAXPES is a surface sensitive technique that interrogates the top 10-12 nm of the NPs, we can conclude that Au atoms diffuse towards the surface disrupting the core-shell structure and changing the local environment of Au and Ag atoms. In previous reports, the Au 4f7/2 BE in small alloy NPs (1-6 nm) was found to be larger than for pure AuNPs, and the opposite tendency was found for the Ag 3d5/2 peak, whose BE decreases in alloy NPs compared to pure AgNPs [24]. These considerations might help to understand the BE shift for samples annealed at 120 °C, although we cannot draw a direct comparison between core shell NPs and alloy NPs due to difficulty in obtaining alloy nanoparticles in this size range.

Fig. 3 Experimental survey scans (from bottom to top) of AuNP annealed at 60 °C (blue line) and 120 °C (orange line), Au@AgNP annealed at 60 °C (yellow line) and 120 °C (purple line), Au@Ag@AuNP annealed at 60 °C (green line) and 120 °C (light blue line). The spectra from all samples annealed at 120 °C are normalized to the Ag 3d area to facilitate the comparison. The arrows indicate the increase in the relative Au content for samples annealed at higher temperature.
Table 2. Surface metal composition of samples annealed at different temperatures.
In addition to the results concerning surface elemental composition obtained from the survey spectra, we analyzed the S 1s HAXPES spectra to obtain further information on the NPs surface state. We can notice that a few peaks are common to all S 1s HAXPES spectra, including commercially available AuNPs and sulfur-terminated ITO/quartz substrates; these peaks can be easily assigned to sulfur-metal species (2469.5 eV), unbound thiols that can originate from unreacted surface dithiols (2471.3 eV) [25], and oxidized sulfur species (disulfides and sulfate species, 2773-2478 eV) [26,27]. Interestingly, in case of S 1s HAXPES spectra for silver containing NPs annealed at 60 °C, we can notice the presence of an additional peak at a lower BE (2468.5 eV). The origin of this peak can be attributed to sulfur bound to surface defects states in silver, most likely charged surface atoms [28] or crystal defects on the NPs surface [29]. This low BE peak is found to disappear for samples annealed at 120 °C, suggesting a thermal migration of Ag defects, which is deemed to facilitate the interdiffusion of atoms. Furthermore, for samples annealed at 120 °C, there is an increase in the intensity for the peaks belonging to oxidized sulfur species. The increase in oxidized sulfur species for 120 °C annealed samples is also accompanied by a possible decrease in the order of the self-assembled layer which is suggested by the decrease in intensity and broadening of the C 1s peak [30] (data not shown).
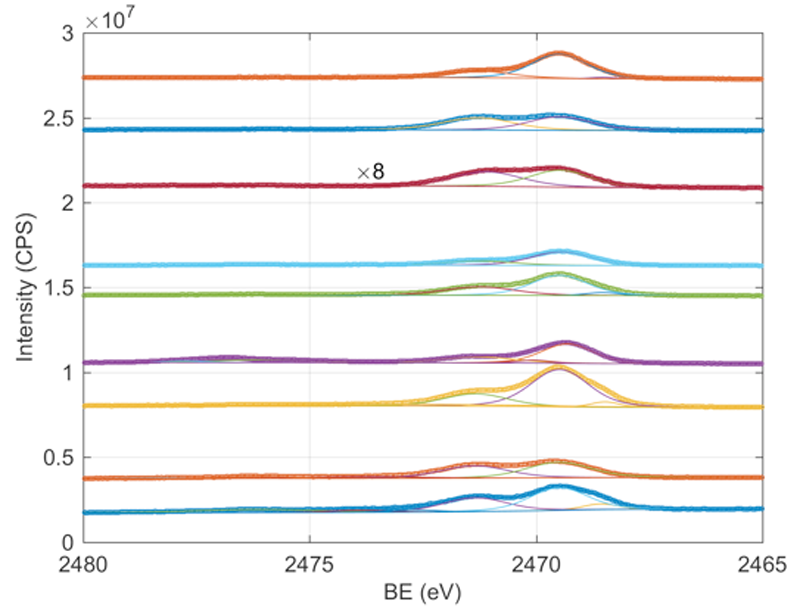
Fig. 4 Experimental S 1s HAXPES spectra (from bottom to top) of AuNP annealed at 60 °C (blue circles) and 120 °C (orange circles), Au@AgNP annealed at 60 °C (yellow circles) and 120 °C (purple circles), Au@Ag@AuNP annealed at 60 °C (green circles) and 120 °C (light blue circles), sulfur-terminated ITO/quartz substrate (dark red circles), commercially available AuNPs annealed at 60°C (blue circles) and AgNPs annealed at 60°C (orange circles). The spectra from all samples annealed at 120 °C are normalized to the Ag 3d area to facilitate the comparison. Each experimental spectrum is decomposed in its main contributions obtained from best fit (solid lines).
Conclusions and challenges
The effect of low temperature annealing on core-shell bimetallic nanoparticles has been investigated by HAXPES. A shift in the BE for Au 4f7/2 and Ag 3d5/2 peaks accompanied by a change in the surface elemental composition for samples annealed at 120 °C suggests that interdiffusion takes place at the Au/Ag interface. In order to characterize further this thermal alloying process, in-situ temperature dependent XPS measurements can be beneficial. Furthermore, a comparison with silver-gold alloy NPs and bulk silver-gold alloy might help to clarify the phenomenon observed in this report. In addition, to exclude any interfering effect from the surfactant and the supporting substrate, surfactant-free NPs arrays and non-supported NPs shall be prepared and characterized.
References
[1] S. D. Standridge, G. C. Schatz, and J. T. Hupp, Langmuir 25, 2596 (2009).
[2] K.-S. Lee and M. A. El-Sayed, J. Phys. Chem. B 110, 19220 (2006).
[3] F. Pincella, K. Isozaki, T. Taguchi, Y. Song, and K. Miki, J. Nanosci. Nanotechnol. 15, 1171 (2015).
[4] W. Hou, Z. Liu, P. Pavaskar, W. H. Hung, and S. B. Cronin, J. Catal. 277, 149 (2011).
[5] W. H. Hung, M. Aykol, D. Valley, W. Hou, and S. B. Cronin, Nano Lett. 10, 1314 (2010).
[6] A. Desireddy, C. P. Joshi, M. Sestak, S. Little, S. Kumar, N. J. Podraza, S. Marsillac, R. W. Collins, and T. P. Bigioni, Thin Solid Films 519, 6077 (2011).
[7] K. Isozaki, T. Ochiai, T. Taguchi, K. Nittoh, and K. Miki, Appl. Phys. Lett. 97, 221101 (2010).
[8] F. Pincella, Y. Song, T. Ochiai, K. Isozaki, K. Sakamoto, and K. Miki, Chem. Phys. Lett. 605-606, 115 (2014).
[9] D. M. Mott, D. T. N. Anh, P. Singh, C. Shankar, and S. Maenosono, Adv. Colloid Interface Sci. 185-186, 14 (2012).
[10] C. Shankar, A. Dao, P. Singh, K. Higashimine, F. M. Mott, and S. Maenosono, Nanotechnology 23, 245704 (2012).
[11] S. Nishimura, A. Dao, D. Mott, K. Ebitani, and S. Maenosono, J. Phys. Chem. C 116, 4511 (2012).
[12] Y. Yamashita, S. He, H. Yoshikawa, S. Ueda, K. Kobayashi, T. Lohmüller, and J. P. Spatz, Int. J. Nanosci. 8, 181 (2009).
[13] T. Shibata, B. A. Bunker, Z. Zhang, D. Meisel, C. F. Vardeman, and J. D. Gezelter, J. Am. Chem. Soc. 124, 11989 (2002).
[14] O. Malis, C. Byard, D. Mott, B. N. Wanjala, R. Loukrakpam, J. Luo, and C. J. Zhong, Nanotechnology 22, 025701 (2011).
[15] B. N. Wanjala, J. Luo, B. Fang, D. Mott, and C.-J. Zhong, J. Mater. Chem. 21, 4012 (2011).
[16] G. Frens, Nature 241, 20 (1973).
[17] Y.-K. Park and S. Park, Chem. Mater. 20, 2388 (2008).
[18] N. R. Jana, Analyst 128, 954 (2003).
[19] T. Dadosh, Mater. Lett. 63, 2236 (2009).
[20] T. Ochiai, K. Isozaki, F. Pincella, T. Taguchi, K. Nittoh, and K. Miki, Appl. Phys. Express 6, 102001 (2013).
[21] U. T. D. Thuy, K. Sakamoto, S. Nishiyama, S. Yanagida, N. Q. Liem, and K. Miki, Langmuir 31, 13494 (2015).
[22] B. N. Wanjala, J. Luo, R. Loukrakpam, B. Fang, D. Mott, P. N. Njoki, M. Engelhard, H. R. Naslund, J. K. Wu, L. Wang, O. Malis, and C. J. Zhong, Chem. Mater. 22, 4282 (2010).
[23] A. B. Smetana, K. J. Klabunde, and C. M. Sorensen, J. Colloid Interface Sci. 284, 521 (2005).
[24] M.-J. Kim, H.-J. Na, K. C. Lee, E. A. Yoo, and M. Lee, J. Mater. Chem. 13, 1789 (2003).
[25] D. G. Castner, K. Hinds, and D. W. Grainger, Langmuir 12, 5083 (1996).
[26] H. Peisert, T. Chassé, P. Streubel, A. Meisel, and R. Szargan, J. Electron Spectros. Relat. Phenomena 68, 321 (1994).
[27] J. A. Rodriguez, J. Dvorak, T. Jirsak, G. Liu, J. Hrbek, Y. Aray, and C. Gonzalez, J. Am. Chem. Soc. 125, 276 (2003).
[28] M. Ramstedt and P. Franklyn, Surf. Interface Anal. 42, 855 (2010).
[29] S. George, S. Lin, Z. Ji, C. R. Thomas, L. Li, M. Mecklenburg, H. Meng, X. Wang, H. Zhang, T. Xia, J. N. Hohman, S. Lin, J. I. Zink, P. S. Weiss, and A. E. Nel, ACS Nano 6, 3745 (2012).
[30] T. M. Willey, A. L. Vance, T. Van Buuren, C. Bostedt, L. J. Terminello, and C. S. Fadley, Surf. Sci. 576, 188 (2005).
ⒸJASRI
(Received: June 21, 2016; Early edition: October 25, 2016; Accepted: December 12, 2016; Published: January 31, 2017)
