





Volume4 No.1
SPring-8 Section A: Scientific Research Report
量子ドットのサイズ選択光エッチング反応過程における構造変化の解明
Analysis on Structural Changes of Quantum Dots during Size-selective Photoetching
a大阪大学大学院工学研究科, b京都工芸繊維大学, c(公財)高輝度光科学研究センター
aOsaka University, bKyoto Institute of Technology, cJASRI
- Abstract
-
直径が10 nm前後の半導体ナノ粒子(量子ドット)は、サイズの減少と共にバンドギャップエネルギーが増大する量子サイズ効果や、常温での蛍光発光など特異な性質を有する。我々は、サイズに応じて光吸収波長が変化する量子ドットの特性を利用し、光による粒径制御法「サイズ選択光エッチング法」を開発した。硫化カドミウム(CdS),テルル化カドミウム(CdTe)ナノ粒子について、単色性の高いナノ粒子溶液を得ることに成功している[1-3]。透過型電子顕微鏡観察の結果は、CdS、CdTeの何れも光エッチング反応による粒径・粒径分布の減少を示しているが、反応途中の光学スペクトル変化や反応速度が両者で全く異なっている。CdSとCdTeナノ粒子の光エッチング反応過程の違いを明らかにすることによって、反応効率を改善したり、同手法を他の種類の半導体に適用したりする際に、非常に重要な知見が得られると期待される。
キーワード: 半導体ナノ粒子、サイズ選択光エッチング、小角X線散乱法
背景と研究目的:
半導体ナノ粒子は、粒径の減少とともにバンドギャップが変化するという特異な性質をもつ材料として知られている。その中でも、CdSeやCdTeなど一部の半導体ナノ粒子は、常温で蛍光発光を示し、バイオイメージングのための蛍光色素や、液晶ディスプレイの波長変換材料として利用されている。粒子表面や内部の欠陥準位が少ないとき、バンド端遷移によって発光するため、同じ材料でありながら粒子サイズによって発光波長を細かく制御することが可能である。これは他の有機・無機蛍光体には見られない半導体ナノ材料最大の特徴である。
粒径と粒径分布を制御する方法としては、合成時の反応条件を精密制御することによって核発生・核成長をコントロールする手法が一般的である。しかしながら、例えば発光波長を5 nm精度でつくりわけることは、最新の洗練された合成手法を用いても困難である。そこで我々のグループは、光を使用した全く新しいサイズ制御法を開発した。サイズ選択光エッチング法は、光励起した半導体ナノ粒子から電子のみを外部に取り出し、粒子内に残ったホールの酸化力によってナノ粒子を自己酸化溶解する手法である(図1)。反応の進行とともに粒径が減少し、量子サイズ効果によってバンドギャップが増加する。単色光を照射した場合は、照射した光を吸収できなくなり、反応が自動的に停止する。
つまり、照射光波長に応じたバンドギャップをもつ半導体ナノ粒子が、単分散な状態で得られるのが本手法の特徴である。以前からCdSナノ粒子については光エッチング反応に成功していたが[1,2]、近年はCdSよりバンドギャップの小さいCdTeナノ粒子のエッチングに成功し、可視光の広い範囲で蛍光発光する半導体ナノ粒子を作り出せるようになった[3]。蛍光スペクトル半値幅が20 nm程度のナノ粒子を、2 nmのピーク波長精度で得ることに成功した。反応後のナノ粒子溶液を限外ろ過によって脱塩処理し、TEM観察を行ったところ、確かに平均粒径の減少や単分散化が見られた。しかしながら、後述するように反応中のスペクトル変化の様子がCdSの場合と異なっていることや、十分な反応時間が経過し、ほとんど単色の蛍光スペクトルが得られた際にも長波長側の吸収ピークがかなり残るという点において、漸次的な溶解反応であるCdSの場合とは異なるメカニズムで反応している可能性があった。そこで、反応中のナノ粒子の粒径分布を、反応溶液から取り出すことなく即座に分析できる手法として、小角X線散乱(SAXS)に注目し、SPring-8で光エッチング反応を行いながら、反応途中の溶液を直ちに測定する実験を試みた。

図1.サイズ選択光エッチングの模式図(a),(b)および同手法によって精密にサイズ制御されたCdTeナノ粒子の蛍光発光
実験:
既報に従い、水溶液中に分散したCdS、CdTeナノ粒子の合成を行い、それぞれヘキサメタリン酸(HMP)で修飾されたCdSナノ粒子と、メルカプト酢酸(TGA)によって修飾されたCdTeナノ粒子を得た[4]。なお、CdTeナノ粒子はバンド端発光を示し、その発光量子収率30%程度であった。
CdTeナノ粒子の光エッチングは、以下の手順で行った。まず、合成したTGA修飾CdTeナノ粒子に水酸化カリウム(KOH)および水酸化カドミウムの析出抑制を目的として塩化アンモニウム(NH4Cl)を加え、TeベースでのCdTe濃度が0.4675 mM、KOH 400 mM、NH4Cl 100 mMとなる溶液を調製した。この溶液に酸素をバブリングしながら撹拌し、300 Wキセノンランプより生成した580 nm、6.5 mW/cm2の単色光を照射した。反応中、何度かにわたって溶液をサンプリングし、φ = 2 mmの石英キャピラリー(Mark-tubes)に入れてSAXS測定を行った。HMP修飾CdSナノ粒子の光エッチングもCdTeと同様に行ったが、エッチング溶液の組成はCdS 1 mM、メチルビオロゲン(MV2+) 0.1 mMであり、照射光波長は450 nmであった。
SAXS測定はBL40B2ビームラインに設置された透過型小角散乱ユニットを使用し、カメラ長1140.8 mm、波長1.0 Åの条件下、イメージングプレート検出器(RIGAKU製, R-AXIS VII)を用いて散乱光を記録した。得られたデータは剛体球を仮定した散乱モデルによってフィッティング解析を行った。解析にはIgor Pro 6上で動くマクロプログラムを用い、溶液中に粒径の異なる10種類のナノ粒子が含まれていると仮定して、計算値が測定データに合致するよう各粒径をもつナノ粒子の存在比を最適化した(図2)。なお、各測定の前にナノ粒子以外のイオンを含む水溶液から散乱パターンを得ることでバックグラウンド情報を得て、測定データから減ずることで、ナノ粒子のみからの散乱情報を選択的に得るよう工夫した。
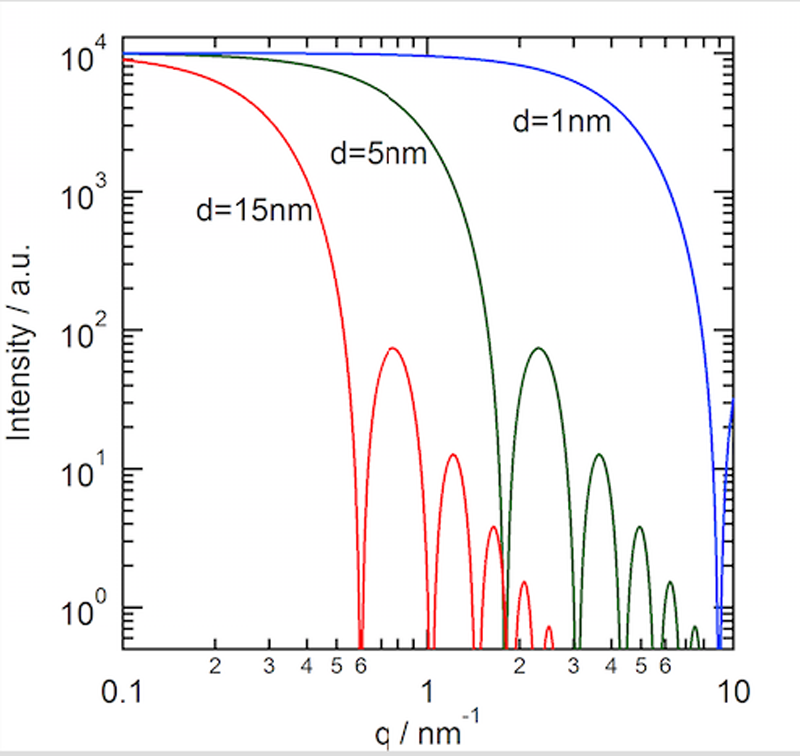
図2.剛体球を仮定した際に得られる粒径ごとの散乱プロファイル。
結果および考察:
CdSナノ粒子の光エッチング反応時の吸収スペクトル変化を図3に示す。バルク状態のCdSは2.4 eV(516 nm)のバンドギャップをもつ半導体であり、比較的粒径の大きい合成直後のCdSナノ粒子はそれに近い吸収端を有する。溶液中で450 nmのモノクロ光照射を行うことによって、吸収端はモノクロメーターの波長幅を考慮した際に妥当と考えられる、440 nm付近までシフトした。また、粒径の減少に伴ってCdS結晶の物質量が減少するため、全体的に光吸収が小さくなる様子が見られた。
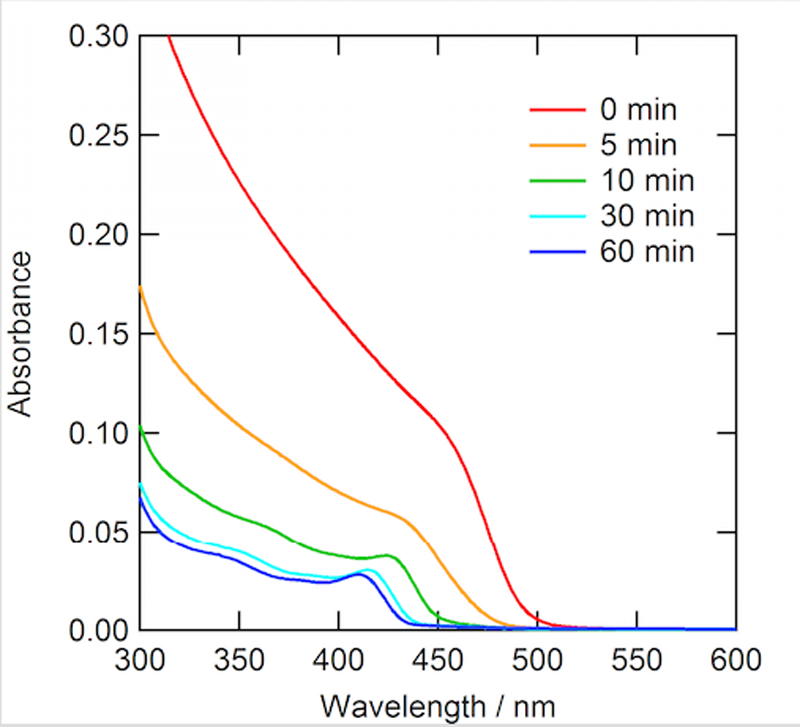
図3.450 nm単色光照射によるCdSナノ粒子の光エッチング反応時の吸収スペクトル変化(同条件ではあるが、SAXS測定を行ったサンプルではないことに留意)
CdSナノ粒子のエッチング過程をSAXSによって解析した。図4(a)はエッチング反応前後の散乱強度の変化である。反応前は10000程度であった散乱強度が、20分の時点で1000程度まで減少していることは、水と異なる電子密度をもつ不溶な物体が、光酸化によって溶解したことを示している。また、散乱ベクトルqの小さい領域の曲線がはっきりと表れていることから、ナノ粒子の粒径分布が単分散に近くなっていることも示唆される。粒径分布解析を行った結果、図4(b)に示すように光エッチング反応前のナノ粒子には、粒径が20 nm前後の大きな構造体が含まれていることが明らかになった。これは、イオン性のナノ粒子同士が水溶液中で完全には独立して存在していないことを示唆している。


図4.CdSナノ粒子の光エッチング反応前後の散乱強度変化(a)と剛体球を仮定してフィッティングを行って得られた、各反応時間における粒径分布(b)。
続いて、光エッチング反応を行うことによって、4–8 nmの範囲で多分散であったCdSナノ粒子が、4 nm程度の単分散なナノ粒子に変化していく様子が観測された。しかしながら、その変化の様子は粒径の漸次的なシフトではなく、大粒径ナノ粒子の消失と考えられるものであった。光溶解によって一時的に配位子が外れることによって不安定化したナノ粒子が一気に溶解するような、瞬時の反応プロセスの存在を示唆しており、従来考えていた漸次的な粒径変化を伴うゆっくりとした酸化溶解反応だけでは説明できない現象が確認された。とりわけ4 nmの直径をもつ粒子の数は反応前後でそれほど変化していないため、4 nmのナノ粒子を残し、これより大きなナノ粒子が完全に消失したと考えるのは自然な見方ではある。しかしながら、これが事実であれば吸収スペクトルは長波長側の吸収を残したまま次第に減少するような変化を見せるはずであり、図3に見られる量子サイズ効果による明確な吸収端のブルーシフトと矛盾する。最初から20 nm程度の大きな構造が含まれていたことは、HMPによる静電的な安定化がそれほど強くは働いていないことを示唆している。その一方でTEM観察によるとこの粒径のナノ粒子が含まれていないため、ナノ粒子の結晶同士が凝集しているのではなく、あくまで溶液中において、配位子を含む形で接近していると考えられる。光エッチング反応中に起こる粒子近傍のイオン強度変化は、ナノ粒子を一層不安定にし、ついには近傍のナノ粒子との融合を引き起こす。粒径が増加したナノ粒子は再度光溶解を受け、この繰り返しが全粒子数の大幅な減少を引き起こしたと考えることができる。
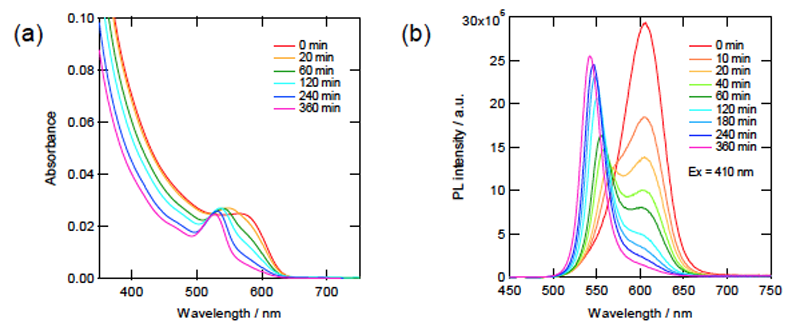
図5.580 nm単色光照射によるCdTeナノ粒子の光エッチング反応時の吸収(a)および蛍光スペクトル(b)の変化(蛍光スペクトル測定時の励起波長は410 nm)。
図5にTGA修飾CdTeナノ粒子の光エッチング反応時の吸収(a)および蛍光スペクトル(b)の変化を示す。30分ほどで完了していたCdSナノ粒子の場合と比べて反応速度はかなり遅く、360分が経過してもまだ照射光波長における吸収が完全には消失していない。蛍光スペクトルのピークは反応初期において完全に二分割され、漸次的な変化ではなく瞬間的な粒径減少を示唆する結果となった[3]。
CdTeナノ粒子の光エッチング反応についてSAXSによる反応中の粒径分布解析を行ったところ、CdSナノ粒子の場合とは全く異なった結果が得られた(図6)。反応の初期段階でわずかに粒子数が減少して以降、蛍光スペクトルが十分に変化した240分が経過した時点でも、粒径分布の変化が全くといっていいほど見られなかった。光学的な変化が起こっていることを踏まえてこの理由を考えると、CdTeナノ粒子が光酸化を受けた際、溶解することなく水酸化カドミウムなどのシェル層となって粒子表面に残存している可能性が挙げられる。CdSと比べて極端に遅い反応速度も、このシェルによる妨害効果によって説明できる。なお、TEM観察時には限外ろ過による脱塩処理を行ったため、化学平衡によってこれらのシェルが溶解し、CdTeコア本来の姿が観察されたものと考えられる。なお、TGA修飾CdTeナノ粒子についても光エッチング反応前から直径20 nmに分布が現れたが、その存在比はHMP修飾CdSナノ粒子の場合と比較すると数分の一であり、チオール基による安定化がHMPによる静電的安定化と比較して、ナノ粒子を溶液中に分散させるという意味で効果的であることが伺える。しかしながら、それらは光エッチング反応によってほとんど数を減らすことがなく、最後まで一定の割合で含まれていた。
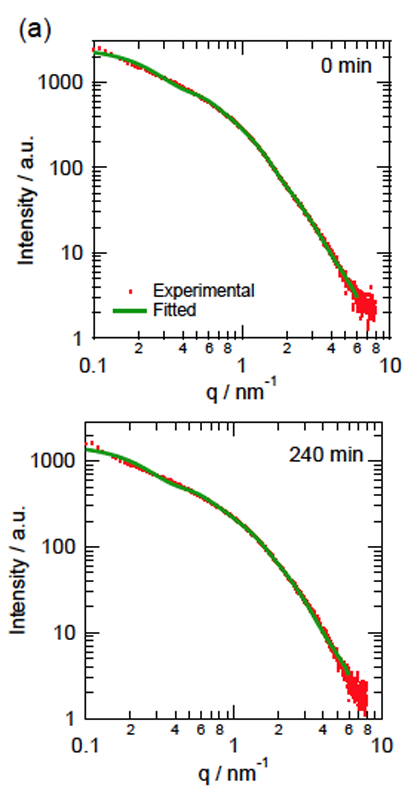
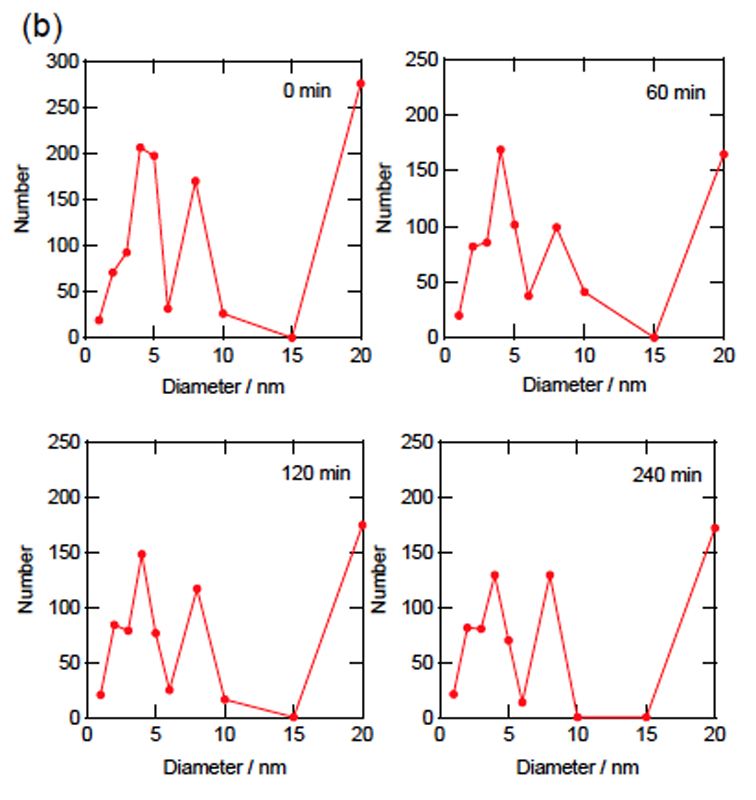
図6.CdTeナノ粒子の光エッチング反応前後の散乱強度変化(a)と剛体球を仮定してフィッティングを行って得られた、各反応時間における粒径分布(b)。
今後の課題:
水溶性CdS、CdTeナノ粒子のサイズ選択光エッチング反応の反応過程で起こる粒径分布変化を、溶液から取り出すことなく解析することに成功した。放射光施設でナノ粒子の光化学反応を行い、時間を置かずに測定するという一連の動作に滞りはなく、新鮮なサンプルを用いて、複雑な前処理を必要とする電子顕微鏡観察では得られない興味深い情報が得られたことは、一定の成果と言える。しかしながら、光エッチングのメカニズムを深く追求したのが初めてであったことや、水溶液中で合成されたナノ粒子が部分的に凝集していたことによって、解析が困難になり、一度の実験で明確な結論を出すことができなかった。また、我々の予想に反し、明確な吸収端シフトが見られるCdSナノ粒子の光エッチングについても、SAXSから得られたデータと光学スペクトル変化の間に差異が見られた。これは、絶縁体である配位子を含んだ形で部分的な凝集が起こった際、その塊が複数の粒子の集合体と見なせるか、1つの大粒径の粒子と見なせるかが、エネルギーバンドの状態によって生じる光学的変化と、小角散乱との間で異なることが一つの原因であると予想される。ナノ粒子の配位子や濃度に注意を払い、溶液中での凝集を一定以下に抑制することによって、解析を妨害する因子を排除する必要がある。これらの事実を含めて再度実験を行うことで、光エッチングの反応機構に関して何らかの結論が得られる可能性は十分にある。
参考文献:
[1] M. Miyake, T. Torimoto, T. Sakata, H. Mori and H. Yoneyama, Langmuir, 15, 1503-1507 (1999).
[2] T. Torimoto, H. Nishiyama, T. Sakata, H. Mori and H. Yoneyama, J. Electrochem. Soc., 145,1964-1968 (1998).
[3] T. Uematsu, H. Kitatjima, T. Kohma, T. Torimoto, Y. Tachibana and S. Kuwabata, Nanotechnology, 20, 215302-215310 (2009).
[4] A. L. Rogach, L. Katsikas, A. Kornowski, D. S. Su, A. Eychmuller and H. Weller, Ber. Bunsenges. Phys. Chem., 100, 1772-1778 (1996).
ⒸJASRI
(Received: July 21, 2015; Early edition: September 25, 2015; Accepted: December 11, 2015; Published: January 25, 2016)
