





Volume3 No.2
SPring-8 Section B: Industrial Application Report
水質分析用ボルタンメトリ法装置に用いる作用電極の表面状態解明-転換電子収量XAFS等の適用可能性
Analysis of the Chemical State of Metals on the Surface of Working Electrode for Stripping Voltammetry – Application of Conversion Electron Yield X-ray Absorption Fine Structure Measurement
a京都大学, b大阪産業大学, cコージェント・エンバイロンメンタル, d(公財)高輝度光科学研究センター
aKyoto University, bOsaka Sangyo University, cCogent Environmental Ltd., dJASRI
- Abstract
-
ボルタンメトリ法による水質分析は、As, Cd, Hg, Se, Pb等の定量下限がICP-質量分析のそれに近く、かつ装置が安価・携帯型かつ低維持費というメリットがある。一方、同法による測定では、環境試料の測定前後で測定感度やバックグラウンド電流が変化するなどの事象が測定誤差のもととなっている。その背景にはボルタンメトリ法に使用する作用電極の表面状態の動的な変化があると考え、転換電子収量XAFSを適用して検討を行った。電極表面に形成された金の膜厚は1-100 nmの範囲と推定されるがS/N比の十分なスペクトルを得ることができた。膜厚の変化がX線吸収スペクトルのジャンプ高さに反映されていると見られた。一方、XAFSスペクトルから見る限り電極上の金は金属状態で、ボルタンメトリ測定時の測定感度に影響を及ぼす金の塩化物や硫化物の形成は本法では検出できなかった。
キーワード: ボルタンメトリ法、水質分析、電極表面、転換電子収量法、XAFS
背景と研究目的:
ボルタンメトリ法は溶液中でμg/Lあるいはそれ以下の濃度レベルの金属イオンの定量を簡便に行える電気化学的分析法であり、ストリッピング・アナリシスとして分類される一群の手法の一つである。著者らの用いている装置は、測定セルに対極(白金)・参照極(Ag/AgCl)・作用極(本研究ではグラッシーカーボン上に金で薄膜を形成させたものを使用)をセットし、20 mLのプラスチックビーカーに試料と支持電解質を入れて測定を行う。所定のプレーティング電圧を印加する前電解過程によって目的元素を作用極に濃縮して、その後の電位走査により目的元素を溶出させるストリッピング過程で、溶出電流を測定する。ストリッピング後のクリーニング過程で作用電極上の目的元素を残らず追い出し、次の測定が可能になる。
ボルタンメトリ法では溶出電位および溶出電流のピーク高さからサンプル中の元素の定性および定量分析を行う。たとえば著者らの使用している装置での定量下限はAs, Cd, Hg, Se, Pbについてそれぞれ5, 0.5, 0.1, 2, 0.5 μg/L(測定時の電解セル内の濃度として)である。環境水中のこれらの元素についてはわが国では公共用水域において10 μg/L以下(総水銀のみ0.5 μg/L)との環境基準が定まっており、ボルタンメトリ法の検出感度は水質監視の目的には十分であるといえる。また、ボルタンメトリ法の装置はいわゆるJIS K 0102の公定法分析装置に比べ極めて安価である。また電極や装置の電流検出系の改良による分析感度の向上が容易で、装置のエンドユーザーでも少しの工夫で高い感度を得ることが可能であるなど手法としての柔軟性が高いことも本法のメリットである。
一般に分析感度の高い手法は、環境分析においては有利である。環境試料中には分析を阻害する様々な共存物質が含まれることが多いが、分析感度が高い手法であれば試料を大幅に希釈して分析に供することで干渉要因を抑制し、正確な分析値を得ることが可能になる。それでも、ボルタンメトリ法による日常分析で測定誤差が発生する場面は少なくない。要因としては、直接的には溶液攪拌の不均一や電極の電気的接触等の装置の不具合によるもの、ならびに試料中の共存物質や試料中の目的元素の化学的存在形態による測定干渉が多い。ただし、上述の場合も、装置不具合や試料組成の影響で作用電極の表面状態の変化が起こり、結果として測定誤差につながっているケースが多い。また、たとえば塩酸(支持電解質)と目的元素しか含まない溶液を測定する際にも金電極などで感度低下が起こることがあり、その原因として電位走査の結果、作用電極の表面の金の酸化状態あるいは塩化物イオンとの結合が変化したとの解釈が発表されている[1-3]。このような電極表面の状態変化については理論的に、あるいは状況証拠から様々な推測が可能であるが、電極表面状態の直接的な観測によって推測を裏付けた例は著者らの知る限り極めて少ない。
そこで今回、作用電極の表面状態を解明するために表面敏感な元素の存在形態分析法として、X線吸収微細構造(XAFS)分析の一つである転換電子収量法の適用について検討した。X線吸収によって内殻電子を失った原子は緩和過程で蛍光X線もしくはオージェ電子を放出するが、その放出の強度からX線吸収量を逆算できる。転換電子収量法はオージェ電子の観測でXAFS測定を行うが、電子は飛距離が短いため、試料表面にかなり近い領域(数十〜数百nm)のみの情報を与える表面敏感な方法となる。
実験:
図1(a)(b)および(c)に転換電子収量測定に供した作用電極を示す。図2に示すようにボルタンメトリ法では、作用電極(working)、対極(counter)、参照電極(reference)の三電極を使用し、ポテンショスタットが参照電極に対する作用電極の電位を制御する。図1(a)(b)に示すように作用電極は先端を取り外すことができる構造のものを特注した。通常の分析で使う電極に比べるとわずかに太いが、感度や電位走査に対する基本的な応答について通常電極との差はなかった。転換電子収量測定時には図1(c)のように取り外した電極先端部を導電性テープ上に張り付けて使用した。今回使用した装置はCogent Environmental社製のPDV6000plusであった。
図1 転換電子収量測定に供した作用電極
(a)作用電極を分解した状態 (b)電極先端を取り外し (c)転換電子収量法での測定

図2 ボルタンメトリ法による測定装置の概要
(a)測定に用いる電極一式 (b)電解セルに電極と試料をセットした状態
(c)電極をセットした電解セルと電位制御を行う装置本体の接続
図3にボルタンメトリ法における電位走査プログラムを模式化して示す。図3から分かるようにボルタンメトリ法では、分析目的元素を作用電極上に濃集させるプレーティング過程、溶出電流を得るストリッピング過程、電極から目的元素をパージするクリーニング過程の一連の電位走査を経て一つのデータを得て、また次の測定ができる状態になる。作用電極が金薄膜で、試料溶液中に硫黄分や有機物が含まれる場合、プレーティング時に金膜に目的元素のみならず硫黄や有機物が濃縮されて膜を不活化し、それ以降の測定において膜が目的元素をプレーティングする能力が低下することが多い。これに対してクリーニング過程は金膜の酸化と共に不要な付着物(硫黄分など)の浄化を促し測定感度を回復させる働きがあると著者らは考えている。また、この電気的なクリーニング過程に代えて10-1 M HCl酸性の10-4 M KMnO4溶液に金薄膜を短時間、浸漬することで膜の表面のクリーニングを行うことも有効なことがこれまでの研究で明らかになった[4]。
そこで今回は、クリーニング過程で作用電極にかける電位であるclean potential、測定溶液中の硫黄分、10-4 M KMnO4液による洗浄,ならびにより濃度の高い10-2 M KMnO4溶液による洗浄がグラッシーカーボン電極上の金膜に与える影響をXAFSで観測することとした。原子吸光用の金標準溶液を40 mg/Lに希釈した溶液からの電着で金薄膜を形成させた後、次の(1)〜(5)の条件下で、ボルタンメトリ法の測定または電極洗浄操作を行ってから、作用電極の先端部分をXAFS測定に供した。(1) clean potential 600 mVにて25 μg/LのAsを1回測定、(2) clean potential 700 mVにて25 μg/LのAsを7回測定、(3) 支持電解質にNa2Sを添加して硫黄濃度が0.1 mg/Lとなるようにしたものを測定、(4) 測定は行わずに10-4 M KMnO4液にて1回洗浄(5秒間撹拌)、(5) 測定は行わずに10-2 M KMnO4溶液にて5回洗浄。ボルタンメトリ法の測定時の支持電解質は、塩化ナトリウム(SIGMA 社製・ASC 級)14.6 g、酢酸ナトリウム(SIGMA社製・Ultrapure級)2.7 gを超純水に溶解したものに酢酸(和光純薬工業社製・精密分析用)1.2 mLを添加して1 Lにメスアップ、これを20 mL中に10 mL添加した。なお、金薄膜形成に先立ち、グラッシーカーボン電極をサンドペーパーで研磨し、前回使用した金薄膜を剥離した。

図3 ボルタンメトリ法における電位走査プログラム
作用電極上の金薄膜の化学的状態および金の厚みの目安を知るために、金のLIII吸収端におけるXAFS測定を行った。転換電子収量法の検出器としてはBL14B2ビームライン備え付けのHeガス収量XAFS測定装置を大気圧He条件下で使用した。この検出器は、X線吸収量に比例して試料薄膜表面から放出されるオージェ電子によって、試料チャンバー中に発生する電離Heを電極に収集・測定することでXAFSスペクトルを得るものである[5]。分光結晶はSi(311)、19.16-16.3°を0.116°/secで走査した。ボルタンメトリ電極の先端部はあらかじめ取り外せるようにした特注品を使用し(図1)、標準物質としては厚み1±0.2 mmの99.99%の金箔、Au2O3・nH2O、AuCl3・nH2O粉末(高純度化学研究所社製)を使用して、導電性の両面テープで測定チャンバー内に固定して測定に供した。得られたXAFS スペクトルはフリーソフトであるAthenaで解析した。
結果および考察:
表1に(1)-(5)の試料について吸収端における電子収量強度のジャンプ量(edge jump)を示す。光電子やAuger電子の脱出深さを反映して転換全電子収量法の検出深さは試料表層に限られる。このため、1 mm厚の金箔で測定された転換全電子強度のジャンプ量0.425はこの手法で測定されるジャンプ量の飽和値である。一方、金薄膜で測定されたジャンプ量は表1に示すように0.142から0.273までと飽和値の0.425より小さい。さて、例えばYanaseら[6]は本研究と同様な大気圧Heイオン収量XAFSにおいて10 nmから10,000 nmの範囲の既知の厚みを持つSrTiO3薄膜試料のSrおよびTiのK吸収端ジャンプ量δμを測定し、δμ値はある膜厚で飽和値に達するまで、膜の厚さと正の相関を有することを示した。同様なことが本研究でも成立すると考え、今回測定した薄膜試料のジャンプ量が金の膜厚を反映した値とみなして以下に考察を行う。なお、文献[7]によれば、ボルタンメトリ電極上の薄膜厚さは1-100 nm の範囲と推定されている。
表1に示すように、ジャンプ量は、(1),(1X),(2)の600 mV,700 mVでの膜のクリーニングの繰り返しによって大きく低下する傾向は認められなかった。(1)と(1X)のように電極を再研磨・再電着するとジャンプ量に差が出たことから、形成される金の膜厚には統計的変動があるとみられる。KMnO4液による洗浄の繰り返しの結果、(4)と(5)を比べると洗浄回数の多い(5)に若干のジャンプ量低下があったが、(1)と(1X)のような膜厚の統計的変化の範囲とも解釈できる。ジャンプ量は支持電解質にNa2Sを添加した場合においてもっとも小さくなった。従来の研究で、試料溶液中の硫化物イオンは、次の反応式左辺のようにまず、水素化した形で金の作用電極の表面に吸着したのち酸化されることが知られている[8]。ここで反応式中のadは金電極に吸着した状態を示す。
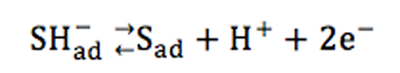
この反応式の右辺の状態で吸着された硫黄はほぼ不可逆的吸着となり、電位走査を行っても容易に外れないと言われる[9]。このことから硫化物イオンを含む環境水中の砒素を金の作用極によりボルタンメトリ分析する場合、硫化物イオンの金へ強固な吸着が、砒素の金電極への電着を妨害し分析感度を不可逆的に低下させると考えられてきた。一方、今回の結果は、試料中に硫化物イオンが含まれる場合に、電極上の金自体が剥離していく可能性を示唆している。金薄膜電極の場合、金が剥離すれば砒素を電着する作用極としての作用が失われることから、これも砒素の分析感度の低下につながる。実際、溶液中に硫化物が存在することで金が溶解しやすくなる現象が知られており、硫化物が金の溶解に触媒的な作用をするためと考察されている[10]。ボルタンメトリの作用極上の金薄膜で同様な現象が起こった可能性は考えられる。
表1 薄膜試料における電子収量強度のジャンプ量
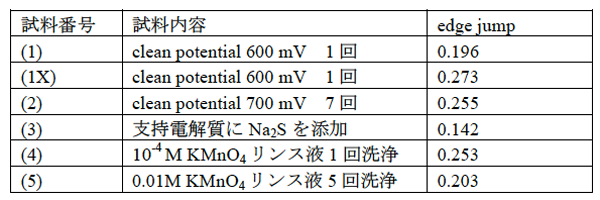
注)表に示す(1X)は(1)で低いedge jump値が出たため値の再現性確認を目的とし同じ電着条件でもう一度作用電極に金薄膜を形成させ、clean potential 600 mVを適用した。
データのうち、標準物質((A) 金箔,(B) Au2O3・xH2O,(C) AuCl3・xH2O)および表1の(2)(3)の薄膜試料のものについて、図4にはXANES領域での規格化したスペクトルを示す。図5にはこれらデータのEXAFS領域でのk3χ(k)を示す。なお、規格化は、吸収端エネルギーE0におけるpre edge lineとpost edge lineの差を基準として行った。吸収端エネルギーE0自体はフリーソフトAthenaのIfefittプログラムの既定の定め方を用いている。すなわち、吸収係数μ(E)の一次微分をとりその第一極大値を選択するが、いわゆるデータのノイズを過って選択しないために、当該極値の低エネルギー側から極値にかけて、エネルギーと共に吸収が増加すること、極値後の高エネルギー側にかけて吸収が順調に低下することを確認する仕様となっている[11]。
図4,5から分かるように薄膜試料についても良好な質のデータが得られ、転換電子収量法適用の成果があった。一方、塩化金などの標準物質と比較すると、電極表面の状態(2)(3)は各種の処理を行ってもいずれも金箔と同様の金属状態で、推測されたような酸化物や塩化物は検出されなかった。
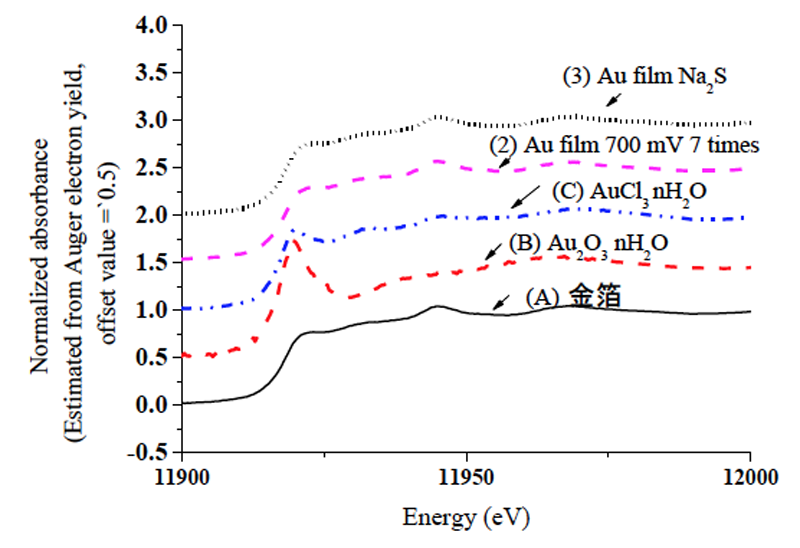
図4 標準試料および薄膜試料の転換電子収量XAFS測定結果:XANES領域でのスペクトル
(縦軸オフセット値=0.5)
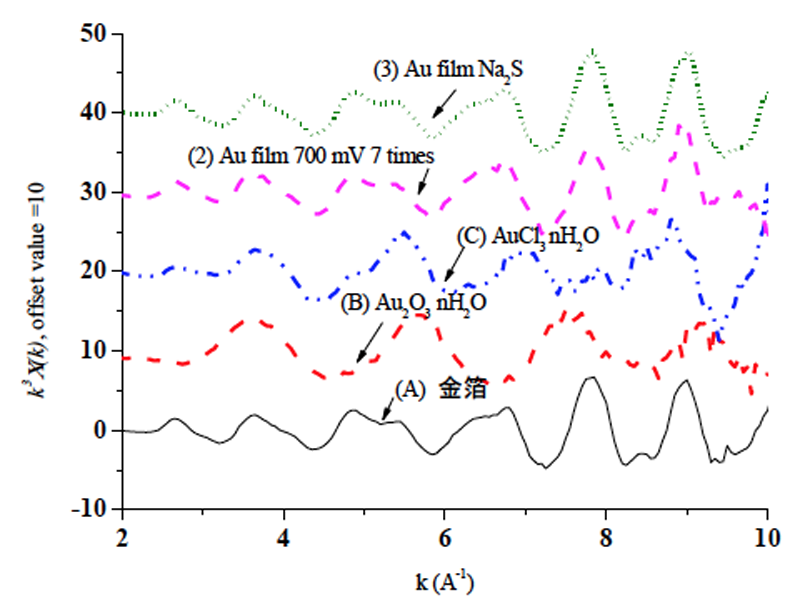
図5 標準試料および薄膜試料の転換電子収量XAFS測定結果:EXAFS領域(k3χ(k))でのスペクトル
(縦軸オフセット値=10)
今後の課題:
ボルタンメトリ法の測定に影響を及ぼす膜の変質は膜のごく表面で起こっており、本方法よりさらに表面敏感な手法での観測を行うことが必要な可能性も考えられる。
参考文献:
[1] B. Lovrecek et al., Electrochimica Acta 26, 1087-1098 (1981).
[2] W. Haiss and J-K. Sass, J. Electroanal. Chem., 431, 15-17 (1997).
[3] P. Salaun and C. V. Berg, Anal. Chem., 78 5052-5060 (2006).
[4] P. Lewtas et al., J.S. Jean, J. Bundschuh, P. Bhattacharya (Eds.), Arsenic in Geosphere and HumanDiseases. CRC Press, The Netherlands, 483-485 (2010).
[5]太田俊明編,X線吸収分光法XAFSとその応用,アイピーシー (2002)
[6] J. Wang, Stripping analysis. VCH Publishers, FL (1985)
[7] E. Yanase et al., Anal. Sci., 15 255-258 (1999).
[8] A. Briceno and S. Chander, J. Appl. Electrochem., 20 506-511 (1990).
[9] Gao S.et al., Langmuir, 8 668-672 (1992).
[10] R. Yu. Bek, L. I. Shuraeva, Russian J. Electrochem., 44 767-771 (2008).
[11] University of Chicago, http://cars9.uchicago.edu/~ravel/software/doc/Athena/html/params/e0.html
ⒸJASRI
(Received: May 8, 2012; Early edition: April 28, 2015; Accepted: June 29, 2015; Published: July 21, 2015)
