





Volume8 No.2
SPring-8 Section B: Industrial Application Report
アクリル共重合体/エポキシ混合系の相分離構造の定量化と機械特性
Quantitative Analysis of Phase Separation Structure and Mechanical Properties of Acrylics Copolymer/ Epoxy Thermo Setting Resin Compositions
リンテック株式会社
LINTEC Corporation
- Abstract
-
アクリル共重合体/エポキシ混合系材料はエポキシ成分の熱硬化による反応誘起型の相分離構造を形成し、アクリル成分のモノマー組成を変えることにより、これが大きく変化する。高輝度放射光を利用して小角X線散乱(SAXS)測定を行い、相分離状態の観察を試み、走査型プローブ顕微鏡(SPM)観察像の画像解析結果と併せて考察した。SAXS 測定結果より一次元相関関数を算出し、相間厚みを求めたところ、SPM より観測される構造を再現する情報を得るに至らなかった。これは分離した相間での電子密度差が小さく、X 線の散乱強度が小さいためと考えられ、特にサブミクロンオーダーの長周期の構造情報が得られなかったものと考えられる。つまりスピノーダル分解による相分離構造が、成分濃度の差の小さい2相により形成されていることが示唆される。
Keywords:相分離、スピノーダル分解、相関長、1次元相関関数、SPM、粒径分布解析
背景と研究目的:
近年、エポキシ樹脂熱硬化材料は接着材料のマトリックスとして多くの工業製品に適用され、特に軽量化が必要とされる有機無機複合機器の技術的進歩を牽引する。半導体装置分野では、素子を接着するためにエポキシ熱硬化材料を応用したフィルム状の接着材が用いられ、厚みの安定性と接着信頼性を両立する。これにより硬度維持のために表面粗度を小さくした薄型素子に対しても充分な接着力を発現し、素子の多積層構造による大容量化を実現している。
しかしエポキシ熱硬化材料が示す物理現象の解明には、その構成分子の構造、組成比、反応性および高次構造などの多くの要素を検討する必要があり、さらにこれらが複雑に関係することから基礎研究分野の研究対象として敬遠される傾向にある。またエポキシ樹脂の示す相分離構造はスピノーダル分解により形成され[1]、接着特性に非常に重要であることが知られている。一方で構造発現現象とそれに起因する物性について定量的に議論した研究が少ない[2]。本研究では、アクリル共重合体/エポキシ熱硬化樹脂混合物が示す相分離構造についての定量化を目的に、SPM 画像について粒径分布を解析し、その構造形成メカニズムについて考察を行った[3]。また同時に SPring-8 の BL19B2 ビームラインにおける小角X線散乱(SAXS)測定および極小角X線散乱(USAXS)測定を行い、相分離状態の定量化を試みた。
実験:
試料 アクリル共重合体(Ac)成分としてメチルアクリレートホモポリマー(MA; Mw = 約 300K)に、エポキシ熱硬化樹脂(Ep)成分としてo-クレゾールノボラック型を主とした4種のエポキシ樹脂とフェノールノボラック型の硬化剤、イミダゾール系硬化促進剤を調整し、Ep 成分が 70 wt% となるよう配合した。これを2-ブタノンに溶解し、塗布および乾燥することにより Ac/Ep 混合フィルムを得た。これをさらに 125°C 1h + 175°C 2h の条件で熱硬化することにより厚さ 40 μm のフィルム状試料を得た(試料名: MA/E4)。熱硬化系の反応性が相構造へ及ぼす影響を検討するために Ep 成分中の硬化促進剤量を1/4倍にしたもの(試料名: MA/E4_0.25)および4倍にしたもの(試料名: MA/E4_4.0)を作製した。また同時に組成物分子構造による相構造変化を検討するため、Ac 成分をメチルアクリレートと2-ヒドロキシエチルアクリレートのランダム共重合体(MH; mol 比 85:15, Mw = 約 300 K)に変えたもの (試料名: MH/E4)、メチルアクリレートとグリシジルメタクリレートのランダム共重合体(MG; mol 比 85:15, Mw = 約 300 K)に変えたもの(試料名: MG/E4)、MH を用い、エポキシ樹脂の種類を2種類にし、ベンゼン環濃度を 4.5×10−3 mol/g から 4.2×10−3 mol/g と小さくしたもの(試料名: MH/E2)を作製した。
形態観察(SPM) SPM 装置 SPA-300HV(日立ハイテクサイエンス社)を用いてタッピングモード(共振周波数: 300–310 kHz)により試料表面 20×20 μm2 の範囲の形状像および位相像を得た。形状像においては、各試料を2-ブタノンに 60 h 浸漬し、表面に偏在する Ac 成分および未反応の Ep 成分を溶出した面を観測し、分離した各相に帰属される2つの高さ分布の交点を閾値に2値化処理をした。これにオリジナルソフトを適用して、高さの高い島相ドメインの粒径の分布とその最近接粒子の中心間距離の分布をそれぞれ求め、次式で示す対数正規分布関数で回帰した。この際、粒径と最近接粒子間距離の平均値(m)および最大頻度を示す最頻値(p)をそれぞれ求め、併せて島相ドメインの面積割合を算出した。また併せて画像解析ソフト Image-J (アメリカ国立衛生研究所(NIH)オープンソースソフトウェア)を用いて FFT 処理を行い、構造周期性と相関長を求めた[4]。
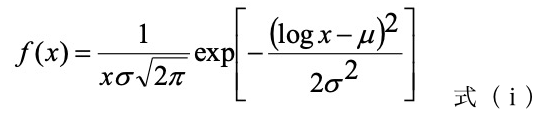
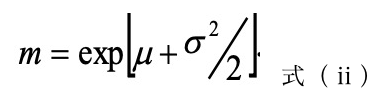
X線散乱(nSAXS および USAXS 測定) SPring-8 BL19B2 ビームライン高輝度放射光を利用して、照射エネルギー 24 keV (波長 0.0516 nm)で2次元検出器(PILATUS-2M)を用いて測定した。カメラ長 3,066 mm (SAXS)の条件では試料直前に直径 1.0 mm のアパーチャを設置し、カメラ長 41,878 mm (USAX)の条件では直径 0.8 mm のアパーチャを設置し、波数 0.00434–3.4 nm−1 の範囲を測定した。なおカメラ長はそれぞれコラーゲンの回折パターンにより較正した。得られた強度プロファイルについては低波数側を Guinier 則により、高波数側を Porod 則により外挿し、式(ⅲ)を用いて全波数領域について逆フーリエ変換し、一次元相関関数 γ(r)を算出した。ここで q、I(q)はそれぞれ測定波数、および散乱強度を示す[5, 6]。さらにγ(r)の極大を与えるrの値から長周期長(l)を、r→0 での立ち上がりを直線近似して、これと γ(r)の極小値を通る水平な直線との交点から相間厚み(d)をそれぞれ求めた。
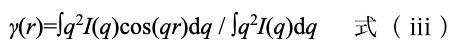
X破断面粗度測定 万能引張試験機 5581 シリーズ (Instron)を用い、250°C での引張強度を測定した後、破断面を SEM(VE-9800, KEYENCE) により観察し、SPM により表面粗度(Ra)を算出した。
結果および考察:
エポキシ硬化系の反応性が及ぼす相構造への影響 SPM により観察した硬化物表面の位相像を図1, a)-1–3 に示す。Ep 成分の硬化促進剤量を増やすとドメイン粒径が小さくなっていることが分かる。画像解析により求めた粒径および最近接粒子間距離の分布を図2に示す。それぞれの観察頻度は実線で示した対数正規分布で再現される。また硬化促進剤量を増やし、反応性が高いものほど、粒径および最近接間距離が小さい。このことから、硬化促進剤量の多い MA/E4_4.0 は成分の分離が充分に進む前に Ep 成分が拡散律速状態になることで硬化反応が終結し構造が決定されると考えられる。

図1. SPM位相像
a)-1 MA/E4_0.25,a)-2 MA/E4,a)-3 MA/E4_4.0
b) MH/E4, c) MG/E4, d) MH/E2
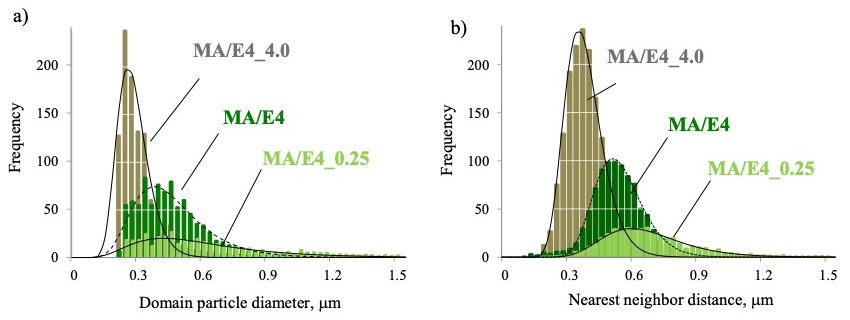
図2. 画像解析結果 (実線および点線;対数正規分布によるフィッティング結果)
a)島相ドメイン粒径 、b)最近接粒子間距離
また対数正規分布に回帰し、求めた m と p の値を表に示す。なお正規分布の場合、両者は同じ値を取る。それぞれの試料において最近接粒子間距離は m とp がほぼ等しい。一方で、粒径は m と p に差があり、硬化促進剤量が少なく、大きな構造を形成するものほどこの差が大きく、正規分布から外れる。物理量が対数正規分布を示す現象として比例効果則に従う現象が挙げられる。本材料系においてもこれに従い、微細に分離したドメインを基にこれらが集積して成長する構造形成過程をとるものと考察される。
これらの 2D-FFT 像を図3, a)– c) に示す。これをみると、中心から離れた位置に同心帯状の明部が存在し、構造に周期性があることが確認される。これらに対し、方位角方向に強度を平均化した1次元強度プロファイルを図3, d) に示す。硬化促進剤量の少ないものほどピーク形状が明瞭となり、周期性が高いことが分かる。ピークを与える波数 (qpeak) から相関長 (lm =2π/qpeak) を求めたものを表に示す。これを見ると、硬化促進剤の多いものほど相関長が短いことが確認され、その値は最近接粒子間距離の平均値(m)の値に対応していることが分かる。これらのことから構造周期性は溶剤不溶の島相の空間的配置によるもので、Ep 成分の熱硬化反応が相分離の駆動力となっていることが分かる。また硬化促進材料の多いものの構造が少ないものの相分離初期段階の構造を模すると考えると、上記の相分離ドメインの大きさと周期性の向上がスピノーダル分解による相分離の終期段階でみられる挙動と一致する。
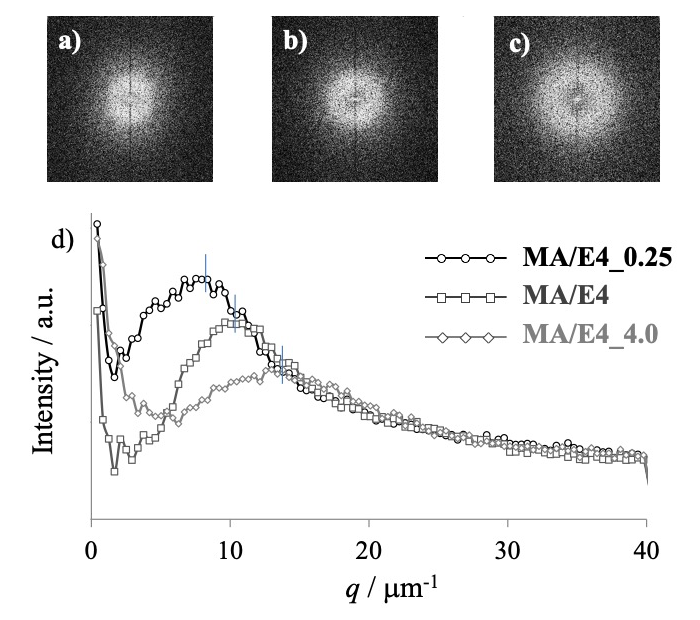
図3. SPM観察像から求めた2D-FFT像
a)MA/E4_0.25、b)MA/E4、c)MA/E4_4.0
および d)1次元強度プロファイル
次に MA/E4 の SAXS および USAXS 測定から得られた散乱強度の1次元プロファイルを図4に示す。他の試料の結果を含め、明瞭なピークは確認されなかった。特に、SPM 観察像から求めた 2D-FFT 像の強度プロファイルでピークの確認された 10 μm−1 (=0.01 nm−1)前後の領域では単調に減少するのみであった。

図4. 小角X線散乱1次元プロファイル(MA/E4)
また、それぞれの SAXS 測定結果より一次元相関関数を算出したものを図5に示す。これを見ると 50 nm 以下の構造において違いが見られるものの 100 nm 以上の構造においては試料間で差が見られない。表に示す相関関数から求めた相間厚み(d)をみると、硬化促進材料を増やし、反応性を高めたものの方が、d の値が大きく、SPM 観察像から求めた最近接間距離と粒径とその傾向も含め絶対値が大きく異なる。また同様に求めた長周期長(l)の値も 126 – 129 nm で、試料間の差も見られず、やはり SPM 観察像の解析結果を再現しなかった。
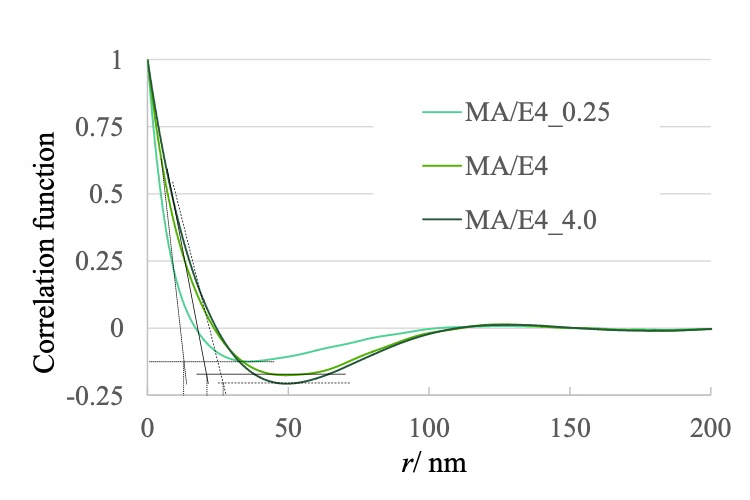
図5. 小角X線散乱より求めた一次元相関関数
表. 組成物分子構造を変えた試料硬化物の画像解析とSAXS解析結果および引張強度
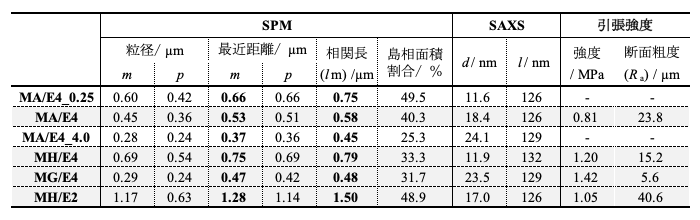
相構造の引張強度への影響と強度発現機構 組成物分子を変えた試料の硬化物表面の SPM 位相像を図1 b)–d) に示す。各組成物を変えることで相分離構造が大きく異なる。表に示した粒径および最近接粒子間距離の結果をみると、先の結果と同様、粒径は m と p の値に差があり、正規分布から大きくはずれることが分かる。また併せて表に示した島相ドメインの面積割合をみると、MA/E4 で 40% であり、設計値と比重から求めたエポキシ成分の体積分率 69% を大きく下回り、エポキシ成分が海相に多く分散した状態であることが分かる。組成を変えた試料はこの値がさらに小さくなるものもあり、この状態が助長されている。
また SAXS 測定結果から求めた d および l は、先の実験同様、SPM で観測される構造情報を再現する結果は得られなかった。X線の散乱測定結果に表れなかった要因として、各相の電子密度の差が小さいことが一因と考えられる。
図6に示す引張強度測定後の破断面をみると、MA/E4 と MH/E2 は表面位相像と似た断面凹凸形状を示し、表に示す Ra の値も lm に応じて変わる。両試料は相境界に沿って破壊が進行していることが示唆される。これに対し MG/E4 は滑らかな断面を示し、Ra が小さい。これは両成分が Ac 中のグリシジル基を介して共有結合を作るため、相境界が曖昧となり両相を貫くことで平滑な破壊面が形成されたものと考えられる。MH/E4 は、破断面の構造が表面相構造より小さく、Ra の値が小さい。このことから部分的に MG/E4 同様、両相を貫き破壊が進行しているものと考えられる。また表から、これらの強度が高いことが分かる。

図6. 引張強度測定後の表面/断面境界像(SEM)
a) MA/E4, b) MH/E4, c) MG/E4 and d) MH/E2
まとめ SPM 観察および粒径解析から硬化促進剤が多いものはドメイン粒径が小さくなる。つまり硬化反応により拡散律速となり、反応の早いものほど相分離が十分成長する前に、構造が形成されている。また粒径の観測頻度は反応性が低いものほど正規分布からはずれ、対数正規分布で再現される。このことから比例効果則に従い、微細に分離したドメインが集積して構造成長していることが示唆される。つまり Ep 成分の硬化が相分離の駆動力となっていることが示される。
しかし SAXS および USAXS の測定および解析からは SPM 観察より得られた構造情報を再現することができなかった。特に USAXS の散乱領域で確認されるサブミクロンオーダーの長周期の構造情報が得られなかった。これは分離した相間での電子密度差が小さく、これによる散乱が小さため、十分解析できなかったものと考えられる。つまりスピノーダル分解により成分が分離するが、その成分の濃度差の小さい2相により構造が形成されていることが示唆される結果となった。このことは、従来から考えられていた構成組成の割合が異なる相により構造が形成されているのではなく、熱硬化成分の共有結合密度に差が生じ、これが相構造を形成している可能性を示すものである。また明確に成分が分離されていないものの方が、各相内部に破壊が進展することで、強度が高くなる結果となった。この結果は、概報される液状エポキシ材料を用いた研究結果[7]と異なる。本研究では、多くの工業製品で汎用される “固形” エポキシ材料とアクリル共重合体の混合物が示す相分離構造を対象とし、その溶融粘度が高いために相形成時の運動性が低いことが要因として挙げられる。エポキシ系材料系の相分離形成メカニズムの新たな事例と認識する。
今後の課題:
SPM 測定から、表面の弾性率分布を確認し、各相での共有結合状態を考察する。
参考文献:
[1] S. Inada et al., J. Network Polym. Japan, 25, 13 (2004).
[2] K. Yamanaka, and T. Inoue, Polymer, 30, 662 (1989).
[3] I. Iichikawa, Doctor Thesis, Tokyo Institute of Technology (2015).
[4] W. S. Rasband, ImageJ, N.I.H., Bethesda, Maryland, USA, (http://imagej.nih.gov/ij/), 1997-2012.
[5] C. G. Vonk, and G. Kortleve, Kolloid-Zeitschrift und Zeitschrift für Polymere 220, 19 (1967).
[6] GRストローブル, 「高分子の物理」(付録A),深尾, 他 訳,シュプリンガージャパン.
[7] H. Kishi et al., eXPRESS Polymer Letters, 9, 23 (2015).
(Received: September 26, 2019; Early Edition: May 27, 2020; Accepted: July 6, 2020; Published: August 21, 2020)
